Canine and Feline Epilepsy
Diagnosis and Management
In memory of Roberto Poma (1969–2010), a friend, colleague and gifted clinician, scientist and teacher.

Diagnosis and Management
In memory of Roberto Poma (1969–2010), a friend, colleague and gifted clinician, scientist and teacher.
Animal Health Trust, UK
University of Georgia, USA

CABI is a trading name of CAB International
| CABI | CABI |
| Nosworthy Way | 38 Chauncy Street |
| Wallingford | Suite 1002 |
| Oxfordshire OX10 8DE | Boston, MA 02111 |
| UK | USA |
| Tel: +44 (0)1491 832111 | Tel: +1 800 552 3083 (toll free) |
| Fax: +44 (0)1491 833508 | E-mail: cabi-nao@cabi.org |
| E-mail: info@cabi.org | |
| Website: www.cabi.org |
© L. De Risio and S. Platt, 2014. All rights reserved. No part of this publication may be reproduced in any form or by any means, electronically, mechanically, by photocopying, recording or otherwise, without the prior permission of the copyright owners.
A catalogue record for this book is available from the British Library, London, UK.
Library of Congress Cataloging-in-Publication Data
De Risio, Luisa, author.
Canine and feline epilepsy : diagnosis and management / Luisa De Risio, Simon Platt.
pages cm Includes bibliographical references and index. ISBN 978-1-78064-109-6 (alk. paper)
1. Dogs--Diseases. 2. Cats--Diseases. 3. Epilepsy in animals. I. Platt, Simon R., author. II. Title.
[DNLM: 1. Dog Diseases. 2. Epilepsy--veterinary. 3. Cat Diseases. SF 992.E57]
SF992.E57D47 2014
636.7'089853--dc23
2013042973
ISBN-13: 978 1 78064 109 6
Commissioning editor: Julia Killick Editorial assistant: Alexandra Lainsbury Production editor: Shankari Wilford
Typeset by SPi, Pondicherry, India Printed and bound in the UK by CPI Group (UK) Ltd, Croydon, CR0 4YY
Contents
Seizures are one of the most common neurological conditions encountered in small animal practice. Epilepsy is the most common chronic neurological disease in dogs; it is often associated with dramatic clinical signs, lifelong treatment and potential effects on the animal quality of life and lifespan. The emotional and financial impact of this disease on the pet-owners can be dramatic.
Diagnosis and management of the seizure patient can be challenging. To date, information on various aspects of this topic has been covered in individual chapters in neurology, internal medicine and pharmacology textbooks and in scientific articles. Therefore consultation of numerous publications has been necessary to obtain comprehensive knowledge.
The authors have compiled this textbook in order to provide information on multiple aspects of canine and feline seizures and epilepsy such as pathophysiology, classification, aetiologies and differential diagnoses, epidemiology, diagnostic investigations and emergency and maintenance treatment. Mechanism of action, metabolism and pharmacokinetics, pharmacokinetic interactions and adverse reactions, dosing, monitoring recommendations and efficacy of old and new generation antiepileptic medications are presented in detail. A glossary on pharmacological terminology has been added at the end of the book to help understanding in this area. Extensive referencing has been provided.
Having all of this information available in one textbook should help to improve knowledge on this complex subject and subsequently help veterinarians to improve the care of dogs and cats with seizure activity.
The authors are very grateful to the invited authors, Holger Volk and Fiona James, for their invaluable contribution in their respective area of expertise, Pathophysiology of refractory seizures (Chapter 2) and Electroencephalography (Chapter 11), and to colleagues who have contributed images. We also wish to thank the publisher CABI and particularly our senior editorial assistant, Alexandra Lainsbury.
Due to production requirements the number of colour images is greater in the e-book than in the printed book. The layout of colour images in the printed book has been determined by the publisher’s production.
We hope that this textbook provides useful information for veterinary students, veterinary general practitioners, as well as veterinary interns, residents and specialists in neurology or in disciplines related to neurology (e.g. internal medicine, oncology, surgery, behavioural medicine and pharmacology). The depth of the information provided will allow those who would like a little bit more detail in certain areas hopefully to find this; however, we hope that the book also serves as a practical source for advancing the treatment of the most routine and most challenging seizure cases.
The information provided in this textbook is up to date to the best of the authors’ knowledge at the time of production. The field of veterinary science is rapidly evolving and advances in diagnosis and treatment are likely to occur in the next years. Therefore knowledge would require constant updating to provide optimal management of our patients.
Luisa De Risio Simon Platt October 2013
1 Pathophysiology of Seizure Activity
Simon Platt Bvm&S mrCvS Dipl. ACvim (Neurology) Dipl. ECvN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
Several decades have been devoted to the study of the pathophysiology of epilepsy. Increasing knowledge in the field has only contributed to a partial understanding of the underlying mechanisms. Nevertheless, insight into the pathophysiology of epilepsy and its underlying histological and neurochemical alterations has contributed to rational development strategies of new antiepileptic medications (AEMs). Although various epileptic syndromes in people have been shown to differ pathophysiologically, they apparently share common ictogenesisrelated characteristics such as increased neuronal excitability and synchronicity. Emerging insights point to alterations of synaptic functions and intrinsic properties of neurons as common mechanisms underlying hyperexcitability. Progress in the field of molecular genetics has revealed arguments in favour of this hypothesis as mutations of genes encoding ion channels were recently discovered in some forms of human epilepsy.
Epileptic seizures arise from an excessively synchronous and sustained discharge of a group of neurons. The single feature of all epileptic syndromes is a persistent increase of neuronal excitability. Abnormal cellular discharges may be associated with a variety of causative factors such as trauma, oxygen deprivation, tumours, infection and metabolic derangements. However, no specific causative factors are found in many dogs and cats suffering from epilepsy.
Underlying causes and pathophysiological mechanisms are (partially) understood for some forms of epilepsy, at least in people, e.g. epilepsies caused by disorders of neuronal migration and monogenic epilepsies. For several other types of epilepsy, current knowledge is only fragmentary. This chapter will review several areas that are understood to contribute to the evolution and maintenance of epilepsy. The genetics of epilepsy are discussed in Chapter 6.
The Electrical Basis of Nerve Cell Function
At the most fundamental level, the nervous system is a function of its ionic milieu, the chemical and electrical gradients that create the setting for electrical activity. Therefore, some of the most easily appreciated controls on excitability are the ways the nervous system maintains the ionic environment. An example is the electrical basis of resting membrane potential. Resting potential is set normally so that neurons are not constantly firing but are close enough to threshold so that it is still possible that they can discharge, given that action potential generation is essential to CNS function. The control of resting potential
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
S. Platt
becomes critical to prevent excessive discharge that is typically associated with seizures.
Normally a high concentration of potassium exists inside a neuron and there is a high extracellular sodium concentration, as well as additional ions, leading to a net transmembrane potential of −60 mV (Scharfman, 2007). If the balance is perturbed (e.g. if potassium is elevated in the extracellular space), this can lead to depolarization that promotes abnormal activity in many ways (Somjen, 2002): terminals may depolarize, leading to transmitter release, and neurons may depolarize, leading to action potential discharge. Pumps are present in the plasma membrane to maintain the chemical and electrical gradients, such as the sodium–potassium ATPase, raising the possibility that an abnormality in these pumps could facilitate seizures. Indeed, blockade of the sodium–potassium ATPase can lead to seizure activity in experimental preparations (Vaillend et al., 2002), suggesting a role in epilepsy (Grisar et al., 1992). The sodium–potassium pump is very interesting because it does not develop in the rodent until several days after birth, and this may contribute to the greater risk of seizures in early life (Haglund et al., 1985; Fukuda and Prince, 1992). In addition to pumps, glia also provide important controls on extracellular ion concentration, which has led many to believe that glia are just as important as neurons in the regulation of seizure activity (Duffy and MacVicar, 1999; Fellin and Haydon, 2005). Thus, the control of the ionic environment provides many potential targets for novel anticonvulsants. It is important to bear in mind that seizures, by themselves, can lead to the changes in the transmembrane gradients. For example, seizures are followed by a rise in extracellular potassium, a result of excess discharge. This can lead to a transient elevation in extracellular potassium that can further depolarize neurons. Thus, the transmembrane potential is a control point that, if perturbed, could elicit seizures and begin a ‘vicious’ cycle, presumably controlled by many factors that maintain homeostasis, such as pumps and glia.
The ionic basis of the action potential is another example of a fundamental aspect of neurobiology that can suggest potential mechanisms of seizures. Neurons are designed to discharge because of an elegant orchestration of sodium and potassium channels that rely on chemical and ionic gradients across the cell membrane. Abnormalities in the sodium channel might lead to a decrease in the threshold for an action potential to occur if the method by which sodium channel activation is controlled alters in any way (i.e. sodium channels are activated at more negative resting potentials or sodium channel inactivation is impaired). Indeed, it has been shown that mutations in the subunits of the voltagedependent sodium channels can lead to epilepsy. A specific syndrome, generalized epilepsy with febrile seizures, is caused by mutations in selected genes responsible for subunits of the voltagedependent sodium channel (Meisler et al., 2001). The mutation does not block sodium channels, presumably because such a mutation would be lethal, but they modulate sodium channel function. This concept, that modulation, rather than essential function, is responsible for genetic epilepsies, has led to a greater interest in directing the development of new anticonvulsants at targets that are not essential to, but simply influence, CNS function.
Synaptic Transmission
Research into seizures has gravitated to mechanisms associated with synaptic transmission, because of its critical role in maintaining the balance between excitation and inhibition. As more research has identified the molecular mechanisms of synaptic transmission, it has become appreciated that defects in almost every step can lead to seizures. Glutamatergic and gaminobutyric acid (GABA)ergic transmission, as the major excitatory and inhibitory transmitters of the nervous system, respectively, have been examined in great detail. It is important to point out, however, that both glutamate and GABA may not have a simple, direct relationship to seizures. One reason is that desensitization of glutamate and GABA receptors can reduce effects, depending on the timecourse of exposure. In addition, there are other reasons. GABAergic transmission can lead to depolarization rather than hyperpolarization if the gradients responsible for ion flow through GABA receptors are altered. For example, chloride is the major ion that carries current through GABAA receptors, and it usually hyperpolarizes neurons because chloride flows into the cell from the extracellular space. However, the K+Cl− cotransporters (KCCs) that are pivotal to the chloride gradient are not constant. In development, transporter expression changes, and this has led to evidence that one of the transporters, NKCC1, may explain seizure susceptibility early in life (Dzhala et al., 2005). The relationship of glutamate to excitation may not always be simple either. One reason is that glutamatergic synapses innervate both glutamatergic neurons and GABAergic neurons in many neuronal systems. Exposure to glutamate could have little net effect as a result, or glutamate may paradoxically increase inhibition of principal cells because the GABAergic neurons typically require less depolarization by glutamate to reach threshold. It is surprisingly difficult to predict how glutamatergic or GABAergic modulation will influence seizure generation in vivo, given these basic characteristics of glutamatergic and GABAergic transmission.
Synchronization
Excessive discharge alone does not necessarily cause a seizure. Synchronization of a network of neurons is involved. Therefore, how synchronization occurs becomes important to consider. There are many ways neurons can synchronize. In 1964, Matsumoto and AjmoneMarsan found that the electrographic events recorded at the cortical surface during seizures corresponded to paroxysmal depolarization shifts (PDS) of cortical pyramidal cells occurring synchronously (Matsumoto and Marsan, 1964). These studies led to efforts to understand how neurons begin to fire in concert when normally they do not. Glutamatergic interconnections are one example of a mechanism that can lead to synchronization. Indeed, studies of the PDS suggested that the underlying mechanism was a ‘giant’ excitatory postsynaptic potential, although it was debated widely at that time if this was the only cause (Johnston and Brown, 1984). Thus, pyramidal cells of cortex are richly interconnected to one another by glutamatergic synapses. Gap junctions on cortical neurons are another mechanism for synchronization. Gap junctions allow a lowresistance pathway of current flow from one cell to another, so that coupled neurons are rapidly and effectively synchronized. It was thought that gap junctions were rare, so it was unlikely that they could play a major role, but further study led to the appreciation that even a few gap junctions may have a large impact on network function (Traub et al., 2004). Another mechanism of synchronization involves, paradoxically, inhibition.
Many GABAergic neurons that innervate cortical pyramidal cells, such as the cell type that controls somatic inhibition (the basket cell), make numerous connections to pyramidal cells in a local area. Therefore, discharge of a single interneuron can synchronously hyperpolarize a population of pyramidal cells. As GABAergic inhibition wanes, voltagedependent currents of pyramidal cells become activated. These currents, such as Ttype calcium channels and others, are relatively inactive at resting potential, but hyperpolarization relieves this inhibition. The result is a depolarization that is synchronous in a group of pyramidal cells (Scharfman, 2007).
Some of the changes that develop within the brain of individuals with epilepsy also promote synchronization. Such changes are of interest in themselves because they may be one of the reasons why the seizures are recurrent. These changes include growth of axon collaterals of excitatory neurons, typically those that use glutamate as a neurotransmitter and are principal cells. An example is the dentate gyrus granule cell of hippocampus. In animal models of epilepsy and in patients with intractable temporal lobe epilepsy (TLE), the axons of the granule cells develop new collaterals and the new collaterals extend for some distance. They do not necessarily terminate in the normal location but in a novel lamina, one that contains numerous granule cell dendrites. Electron microscopy has shown that the new collaterals innervate granule cell dendrites, potentially increasing recurrent excitatory circuits. Some argue that recurrent inhibition increases as well as recurrent excitation, but the
S. Platt
fact remains that new synaptic excitatory circuits develop that are sparse or absent in the normal brain (Nadler, 2003; Sloviter et al., 2006). The resultant ‘synaptic reorganization’ not only can support synchronization, potentially, but it also illustrates how the plasticity of the nervous system may contribute to epileptogenesis.
Kindling and Epileptogenesis
Goddard (1967) was the first to describe that periodic stimulation of neural pathways progressively leads to recurrent behavioural and electrographic seizures. Kindling procedures have provided a substrate for the study of the role of enhanced synaptic efficacy in seizure disorders. It is now considered to be a first choice experimental procedure in the study of the potential mechanisms of epileptogenesis. The phenomenon can be evoked in various brain regions, but amygdala kindling is most frequently used in epilepsy research as a model for complex focal (partial) seizures (Fisher, 1989). Although kindling has been shown to be phenomenologically different from other types of plastic changes in the central nervous system, there are many points of similarity between kindling and the process of longterm potentiation (Sutula et al., 1989).
Kindling has been shown to depend upon functional as well as structural changes in glutamatergic synapses. The anticonvulsant effects of glutamate receptor blocking agents like NmethylDaspartate (NMDA) antagonists seem to be at least partly due to their inhibitory effects on in vitro kindling.
ictogenesis
Excitability is a key feature of ictogenesis that may originate from individual neurons, neuronal environment or a population of neurons. Excitability arising from single neurons may be caused by alterations in membrane or metabolic properties of individual neurons (Traub et al., 1996). When regulation of environmental, extracellular concentrations of ions or neurotransmitters is suboptimal, the resulting imbalance might enhance neuronal excitation. Collective anatomic or physiologic neuronal alterations may convert neurons into a hyperexcitable neuronal population. In reality, these three theoretical mechanisms are thought to interact during specific ictal episodes. Each epileptic focus is unique as the differential contribution of these three concepts leading to ictal events is thought to differ from focus to focus.
Excitability arising from individual neurons
Functional and perhaps structural changes occur in the postsynaptic membrane, thus altering the character of receptor proteinconductance channels, thereby favouring development of paroxysmal depolarizing shifts (PDS) and enhanced excitability. Epileptic neurons appear to have increased Ca2+ conductance. It may be that latent Ca2+ channels are used, that the efficacy of Ca2+ channels is increased or that the number of Ca2+ channels is chronically elevated. However, development of burst activity depends on the net inward current and not on the absolute magnitude of the inward current. When extracellular K+ concentrations are increased (as during seizure activity), the K+ equilibrium across the neuronal membrane is reduced, resulting in reduced outward K+ currents. The net current will become inward, depolarizing the neuron to the extent that Ca2+ currents will be triggered. This results in a PDS and a burst of spikes (Dichter and Ayala, 1987).
Excitability arising from neuronal microenvironment
Both functional and structural alterations occur in epileptic foci. The functional changes involve concentrations of cations and anions, metabolic alterations, and changes in neurotransmitter levels. The structural changes involve both neurons and glia. Excessive extracellular K+ depolarizes neurons and leads to spike discharge. During seizures, changes in extracellular Ca2+ (a decrease of 85%) precede those of K+ by milliseconds and Ca2+ levels return to normal more quickly than K+. Glia are able to clear neurotransmitters from the extracellular space and to buffer K+, thus correcting the increased extracellular K+ concentrations that occur during seizures. Epileptic foci may show proliferation of glia that differ however in morphological and physiological properties (Bordey and Sontheimer, 1998). Gliosis will affect glial K+ buffering capacity and hence may contribute to seizure generation.
The epileptic cell population
Collective anatomic or physiologic neuronal alterations might produce progressive, networkdependent facilitation of excitability, perhaps coupled with a decrease of inhibitory influences, e.g. due to selective loss of inhibitory neurons. Mossy fibre sprouting (MFS) is an example of neuronal alterations leading to increased excitability (Cavazos et al., 1991). MFS was demonstrated in patients with refractory temporal lobe epilepsy with hippocampal sclerosis on neuroimaging as well as in numerous animal models of temporal lobe epilepsy (Sutula et al., 1988, 1989). In normal conditions, the dentate granule cells limit seizure propagation through the hippocampal network. However, the formation of recurrent excitatory synapses between dentate granule cells, as is thought to occur after MFS, may transform the dentate granule cells into an epileptogenic population of neurons (McNamara, 1999). Possibly, a vicious cycle develops: seizures cause neuronal death, which results in MFS, which in turn increases seizure frequency.
mechanisms of interictal–ictal Transition
Mechanisms producing signal amplification, synchronicity and spread of activity are likely to be involved in interictal–ictal transitions. In vivo, interictal–ictal transition can seldom be attributed to one theoretical mechanism, but often results from the interaction of different mechanisms.
Nonsynaptic mechanisms
Alterations in ionic microenvironment
Repetitive ictal and interictal activity causes increases in extracellular K+ leading to increased neuronal excitability (Moody et al., 1974). Some neurons are very sensitive to changes in membrane K+ currents, e.g. pyramidal cells in the CA1 region of the hippocampus (Rutecki et al., 1985).
Active ion transport
Activation of the Na+–K+ pump is important for regulation of neuronal excitability during excessive neuronal discharges. Substances like ouabain that block the Na+–K+ pump can induce epileptogenesis in animal models. Hypoxia or ischaemia can result in Na+–K+ pump failure thus promoting interictal–ictal transition. A Cl−–K+ cotransport mechanism controls the intracellular Cl− concentration and the Cl− gradient across the cell membrane, which regulates effectiveness of GABAactivated inhibitory Cl− currents. Interference with this process could cause a progressive decrease in the effectiveness of GABAergic inhibition leading to increased excitability (Engelborghs et al., 2000).
Presynaptic terminal bursting
The amount of transmitter released is related to depolarization of presynaptic terminals. Changes in axon terminal excitability will have effects on synaptic excitation (Engelborghs et al., 2000). Abnormal bursts of action potentials occur in the axonal arborizations of thalamocortical relay cells during epileptogenesis. Since one thalamocortical relay cell ends on a large number of cortical neurons, synchronization can occur, which might play an important role in interictal–ictal transition (Engelborghs et al., 1998a).
Ephaptic interaction
Ephaptic interactions are produced when currents from activated neurons excite adjacent neurons in the absence of synaptic connections. Ephaptic effects are strongly dependent
S. Platt
on the size of the extracellular space. When extracellular space is small, ephaptic interactions are promoted (Traub et al., 1985).
Synaptic mechanisms
Two theoretical mechanisms can occur: decreased effectiveness of inhibitory synaptic mechanisms or facilitation of excitatory synaptic events. Both mechanisms will be discussed below.
Neurochemical mechanisms Underlying Epilepsy
GABA
The GABA hypothesis of epilepsy implies that a reduction of GABAergic inhibition results in epilepsy whereas an enhancement of GABAergic inhibition results in an antiepileptic effect (Wong and Watkins, 1982; De Deyn and Macdonald, 1990; De Deyn et al., 1990). Inhibitory postsynaptic potentials (IPSPs) gradually decrease in amplitude during repetitive activation of cortical circuits. This phenomenon might be caused by decreases in GABA release from terminals, desensitization of GABA receptors that are coupled to increases in Cl− conductance or alterations in the ionic gradient because of intracellular accumulation of Cl− (Wong and Watkins, 1982). In case of intracellular accumulation of Cl−, passive redistribution is ineffective. Moreover, Cl−–K+ cotransport becomes less effective during seizures as it depends on the K+ gradient. As Cl−–K+ cotransport depends on metabolic processes, its effectiveness may be affected by hypoxia or ischaemia as well. These mechanisms may play a critical role in ictogenesis and interictal–ictal transition. Several studies have shown that GABA is involved in pathophysiology of epilepsy in both animal models and patients suffering from epilepsy. GABA levels and glutamic acid decarboxylase (GAD) activity were shown to be reduced in epileptic foci surgically excised from patients with intractable epilepsy and in CSF of patients with certain types of epilepsy (De Deyn et al., 1990).
A reduction of 3H–GABA binding has been reported in human brain tissue from epileptic patients whereas PET studies demonstrated reduced benzodiazepine receptor binding in human epileptic foci (Savic et al., 1996). The degree of benzodiazepine receptor reduction showed a positive correlation with seizure frequency. The GABA receptor complex is involved in various animal models of epilepsy as well. Low CSF levels of GABA were revealed in dogs with epilepsy (Loscher and SchwartzPorsche, 1986). Reduced GAD levels were revealed in the substantia nigra of amygdalakindled rats (Loscher and Schwark, 1985). Significant alterations in GABA and benzodiazepine binding have been shown in the substantia nigra of genetically seizureprone gerbils (Olsen et al., 1985). Mice with a genetic susceptibility to audiogenic seizures have a lower number of GABA receptors than animals of the same strain that are not seizure prone (Horton et al., 1982). Several endogenous (guanidino compounds) and exogenous
(e.g. bicuculline, picrotoxin, penicillin, pilocarpine, pentylenetetrazol) convulsants inhibit GABAergic transmission through inhibition of GABA synthesis or through interaction with distinct sites at the postsynaptic GABAA receptor (De Deyn and Macdonald, 1990; D’Hooge et al., 1996). Convulsant agents that block synaptic GABAmediated inhibition, amplify the dendritic spikegenerating mechanism that involves Ca2+ (Dichter and Ayala, 1987; Fisher, 1989). Synaptic inputs are thought to trigger and synchronize this process throughout a population of cells, which then might result in an epileptic seizure. Several AEMs are GABA analogues, block GABA metabolism or facilitate postsynaptic effects of GABA. However, a study evaluating dosedependent behavioural effects of single doses of vigabatrin in audiogenic sensitive rats, suggests that the antiepileptic properties of vigabatrin not only depend on GABAergic neurotransmission but might also be explained by decreased central nervous system levels of excitatory amino acids or increased glycine concentrations (Engelborghs et al., 1998b).
Glutamate
In rodent models, altering glutamate receptor or glutamate transporter expression by knockout
or knockdown procedures can induce or suppress epileptic seizures (Chapman et al., 1996; Kabova et al., 1999; Chapman, 2000). Regardless of the primary cause, synaptically released glutamate acting on ionotropic and metabotropic receptors appears to play a major role in the initiation and spread of seizure activity (Meldrum, 1994; Chapman et al., 1996; Chapman, 2000). Glutamatergic synapses play a critical role in all epileptic phenomena. Activation of both ionotropic and metabotropic postsynaptic glutamate receptors is proconvulsant. Antagonists of NmethylDaspartate (NMDA) receptors are powerful anticonvulsants in many animal models of epilepsy. Several genetic alterations have been shown to be epileptogenic in animal models.
Glutamate receptors
Studies of epileptiform discharges in hippocampal slices show that the characteristic burst discharge, associated with a ‘paroxysmal depolarizing shift’, is dependent on activation of AMPA receptors for its initial components and NMDA receptors for the later elements (Bengzon et al., 1999; Mazarati and Wasterlain, 1999; Meldrum et al., 1999).
AMPA
AMPA receptor antagonists, either competitive or noncompetitive, are anticonvulsant in rodent models (Rogawski and Donevan, 1999). Thus, altered function of AMPA receptors could contribute to proconvulsant or anticonvulsant effects (Meldrum et al., 1999). Evidence has accumulated that Ca2+permeable AMPA receptors may play a role in epileptogenesis and the brain damage occurring during the prolonged seizures (Rogawski and Donevan, 1999). Because Ca2+permeable AMPA receptors are predominantly expressed in GABAergic interneurons, it is hypothesized that some forms of epilepsy might be caused by reduced GABA inhibition resulting from Ca2+permeable AMPA receptormediated excitotoxic death of interneurons (Rogawski and Donevan, 1999).
NMDA
NMDA receptor antagonists are potent anticonvulsants in many animal models, suggesting a role for these receptors in epileptogenesis (Patrylo et al., 1999). It is known that enhancing NMDA receptormediated excitatory actions (e.g. by lowering extracellular Mg) produces epileptiform activity in experimental models of ‘kindled’ epilepsy (Chapman, 1998, 2000). It has been postulated that NMDA receptors may change after neuronal damage (Rice and DeLorenzo, 1998). New receptors are formed that have either less sensitivity to ambient Mg or more sensitivity to ambient glycine; increased excitability could occur within local circuits where the circuitry itself is not altered (or may occur in addition to circuit alterations) (Meldrum et al., 1999). As it is known that the NMDA receptor is subject to modulation by a variety of endogenous agents, including glycine (as a coagonist with glutamate), polyamines, steroids, neuropeptides (Vezzani et al., 2000b), pH, the redox state of the receptor, and NO, there are many chronic alterations in NMDA receptors that could underlie longterm changes in excitability and, thereby, epilepsy. Presently, there are no data to support changes in any of these regulatory factors in chronic epilepsy, but it is distinctly possible that alterations in one or more of these will be shown to be responsible for one or another form of inherited epilepsy.
Kindling is the most extensively studied animal model of epileptogenesis, and this has demonstrated the unique importance of NMDA receptors in the creation of seizure activity (Bengzon et al., 1999; Meldrum et al., 1999). In kindling, repeated electrical stimuli in the limbic system lead to a progressive increase of seizure susceptibility. When the animal responds to stimuli with generalized convulsions, it has developed a permanent epileptic condition. Activation of NMDA receptors and levels of NMDA receptor function are critical in kindling epilepsy (Bengzon et al., 1999). Selective NMDAreceptor antagonists retard kindling development and can also, at higher doses, have an anticonvulsant effect (Bengzon et al., 1999; Trist, 2000).
S. Platt
Metabotropic receptors
On account of these receptors’ responsibility for regulating glutamatergic and GABAergic neurotransmission, it is not surprising that mGluRs strongly influence the induction, propagation and termination of epileptic activity in the central nervous system (CNS) (Doherty and Dingledine, 2002). Pharmacological studies with mGluR group specific agonists and antagonists provide a relatively clear picture for Group I, with agonists being convulsant and antagonists being anticonvulsant (Meldrum et al., 1999; Doherty and Dingledine, 2002; Sayin and Rutecki, 2003). The picture is more complicated for the Group II and III receptors but anticonvulsant effects have been described for agonists of both these groups (Meldrum et al., 1999).
Glutamate transporters
In addition to receptor abnormalities, glutamate transporters, responsible for the removal of glutamate from the extracellular fluid, have been implicated in epilepsy (Meldrum et al., 1999). In situ hybridization studies have shown that the mRNA responsible for the rat glial glutamate transporter (GLT) is reduced in several brain regions in epilepsyprone rats (Meldrum et al., 1999). GLT ‘knockout’ mice have been bred to provide homozygous mice, in which the GLT protein is not detected. In such mutant mice, glutamate uptake in cortical synaptosomes is 5.8% compared with the wildtype (Meldrum et al., 1999). The mutant mice show spontaneous seizures, with wild running and tonic extension, which is frequently fatal. In chronic seizure models (kindled seizures, spontaneous seizures and genetically epilepsyprone rats), there are numerous reports of increases in extracellular glutamate during seizures (Meldrum et al., 1999). This strongly suggests that in these chronic models there are sustained functional alterations in mechanisms relating to the synaptic release of glutamate or its transport. GLT1 astrocytic expression was reduced in four Shetland sheepdogs with IE (Morita et al., 2005). In these dogs it was suggested that decreased expression of the transporter might be related to development of status epilepticus.
There is not as yet any genetically determined epilepsy syndrome occurring spontaneously in man or mouse that can be ascribed to a primary gene defect involving a glutamate receptor or transporter.
Targets for treatment
In animal models of epilepsy, antagonists acting at NMDA receptors, AMPA receptors or at Group I metabotropic receptors have potent anticonvulsant actions (Meldrum and Chapman, 1999; Rogawski and Donevan, 1999; Chapman, 2000; Moldrich et al., 2003).
NMDA receptor antagonists have been successful in stopping the maintenance phase of selfsustaining status epilepticus (SE) in rats, which suggests that these compounds may have a promising role in the treatment of unrelenting seizure activity such as SE (Mazarati and Wasterlain, 1999). Studies with selective AMPA receptor antagonists have indicated that AMPA receptors are potentially promising anticonvulsant drug targets, but at present this is uncertain (Rogawski and Donevan, 1999).
In genetic mouse models, mGlu1/5 antagonists and mGlu2/3 agonists are effective against absence seizures. Thus, antagonists at Group I mGlu receptors and agonists at Groups II and III mGlu receptors are potential antiepileptic agents, but their clinical usefulness will depend on their acute and chronic sideeffects (Moldrich et al., 2003). Potential also exists for combining mGlu receptor ligands with other glutamatergic and nonglutamatergic agents to produce an enhanced anticonvulsant effect (Moldrich et al., 2003).
The Veterinary Perspective
Idiopathic epilepsy (see Chapter 6) is the most common cause of seizures in dogs (Podell and Hadjiconstantinou, 1997). Low levels of GABA and high levels of glutamate have been detected in the cerebrospinal fluid of epileptic dogs independent of time relation to recent seizure activity (Podell and Hadjiconstantinou, 1997). The glutamate elevations are not related whether the seizures were focal or generalized in character (Podell and Hadjiconstantinou, 1997). These findings may indicate the brains of epileptic dogs are under a state of chronic overexcitation. Although a separate study found that lower CSF GABA concentration was associated with a reduced response to phenobarbital therapy in dogs, there was no association between CSF glutamate and response to this therapy (Podell and Hadjiconstantinou, 1999). However, a negative association was found between CSF glutamate:GABA ratio and response to phenobarbital therapy (Podell and Hadjiconstantinou, 1999). Therefore glutamatemediated mechanisms may be useful targets for anticonvulsant therapy in dogs. Intracerebral microdialysis was used to demonstrate elevation of extracellular levels of glutamate in four Shetland sheepdogs with IE, suggesting an important role in the occurrence of seizure activity (Morita et al., 2005).
Gabapentin (see Chapter 17), a relatively new human anticonvulsant, has been evaluated in dogs refractory to phenobarbitone and potassium bromide with an approximate 50% success rate.
Gabapentin has been shown to modestly decrease glutamate levels in the brain (Errante and Petroff, 2003). Another new anticonvulsant, topiramate (see Chapter 19), produces its antiepileptic effect by several mechanisms, one of which is inhibition of kainitemediated glutamate receptors (Angehagen et al., 2003a). This drug has also been demonstrated to protect neurons from excitotoxic levels of glutamate, potentially preventing brain damage during seizure activity (Angehagen et al., 2003b).
Catecholamines
Abnormalities of CNS catecholamines have been reported in several genetic models of epilepsy. In the spontaneous epileptic rat, dopamine was decreased in the caudate nucleus whereas noradrenaline was increased in the midbrain and brainstem (Hara et al., 1993). Decreased levels of dopamine have been found in epileptic foci of epilepsy patients (Mori et al., 1987). In animal models of absence epilepsy, seizures are exacerbated by dopamine antagonists while alleviated by dopamine agonists (Snead, 1995). These results suggest that decreased dopamine facilitates appearance of seizures by lowering the threshold triggering such seizures. Tottering mice have an absencelike syndrome that is characterized by episodes of behavioural arrest associated with 6 to 7 Hz cortical SW EEG discharges. Selective destruction of the ascending noradrenergic system at birth prevents the onset of the syndrome. Therefore, it has been suggested that the syndrome is caused by a noradrenergic hyperinnervation of the forebrain (Engelborghs et al., 2000). Recent data indicate that the serotonergic system regulates epileptiform activity in a genetic rat model of absence epilepsy as intraperitoneal or intracerebroventricular administration of 8 OHDPAT caused marked and dosedependent increases in number and duration of discharges (Gerber et al., 1998).
Opioid peptides
In experimental studies, opioids and opioid peptides had both convulsant and anticonvulsant properties (Engelborghs et al., 2000). Kappa agonists suppress electrical discharges in an animal model of absence epilepsy (Przewlocka et al., 1995). Peptides with a magonist action induce hippocampal or limbic seizures when administered intraventricularly possibly due to inhibition of inhibiting interneurons. In patients with complex partial seizures, PET studies pointed out that mreceptor density is increased in the temporal cortex (Mayberg et al., 1991).
inflammatory mechanisms Underlying Epilepsy
Over the past 10 years an increasing body of clinical and experimental evidence has provided strong support to the hypothesis that inflammatory processes within the brain might constitute a common and crucial mechanism in the pathophysiology of seizures and epilepsy (Vezzani et al., 2011). The first insights into the potential role of inflammation in human epilepsy were derived from clinical evidence indicating that steroids and other antiinflammatory treatments displayed anticonvulsant activity in some drugresistant epilepsies (Wirrell et al., 2005; Wheless et al., 2007). Additional evidence came from febrile seizures in people, which always coincide with, and are often caused by, a rise in the levels of proinflammatory agents (Dube et al., 2007). Evidence of immune system activation in some patients with seizure disorders, the high incidence of seizures in autoimmune diseases, and the discovery of limbic encephalitis as a cause of epilepsy led to the suggestion that immune and inflammatory mechanisms have roles in some forms of epilepsy (Aarli, 2000; Bien et al., 2007; Vincent and Bien, 2008; Vezzani et al., 2011).
Evidence is emerging that inflammation might be a consequence as well as a cause of epilepsy. Several inflammatory mediators have been detected in surgically resected brain tissue from human patients with refractory epilepsies, including temporal lobe epilepsy (TLE) and cortical dysplasiarelated epilepsy (Choi et al., 2009; Vezzani et al., 2011). The finding that brain inflammation occurred in epilepsies that were not classically linked to immunological dysfunction highlighted the possibility that chronic inflammation might be intrinsic to some epilepsies, irrespective of the initial insult or cause, rather than being only a consequence of a specific underlying inflammatory or autoimmune aetiology. The mounting evidence for a role for inflammatory processes in human epilepsy has led to the use of experimental rodent models to identify putative triggers of brain inflammation in epilepsy, and to provide mechanistic insights into the reciprocal causal links between inflammation and seizures (Vezzani et al., 2011). Experimental studies have shown that seizure activity per se can induce brain inflammation, and that recurrent seizures perpetuate chronic inflammation. Seizureassociated cell loss can contribute to inflammation but is not a prerequisite for inflammation to occur. In addition, models of systemic or CNS infections suggested that preexisting brain inflammation increases the predisposition to seizures, associated with alterations in neuronal excitability and enhanced seizureinduced neuropathology. Additional mechanistic insights into the role of inflammation in seizures and the development of epilepsy have been gained through use of pharmacological approaches that interfere with specific inflammatory mediators and from changes in seizure susceptibility in genetically modified mice with perturbed inflammatory pathways (Campbell et al., 1993; Kelley et al., 1999; Vezzani et al., 2000a; Balosso et al., 2005).
Inflammation consists of the production of a cascade of inflammatory mediators (a dynamic process), as well as antiinflammatory molecules and other molecules induced to resolve inflammation, as a response to noxious stimuli (such as infection or injury), or immune stimulation, and is designed to defend the host against pathogenic threats. Inflammation is characterized by the production of an array of inflammatory mediators from tissueresident or bloodcirculating immunocompetent cells, and involves activation of innate and adaptive immunity. Both innate and adaptive immunity have been implicated in epilepsy, and microglia, astrocytes and neurons are believed to contribute to the innate immunitytype processes that cause inflammation of the brain. The brain has traditionally been considered an immunoprivileged site because of the presence of the blood–brain barrier (BBB), the lack of a conventional lymphatic system, and the limited trafficking of peripheral immune cells. Nevertheless, both the innate and adaptive immune responses are readily evoked within the CNS in response to pathogens, selfantigens, or tissue injury of several aetiologies. Microglia, astrocytes, neurons, BBB endothelial cells, and peripheral immune cells extravasating into brain parenchyma can all produce proinflammatory and anti inflammatory molecules (Ransohoff et al., 2003; Banks and Erickson, 2010).
The contribution of each cell population to brain inflammation depends on the origin (for example, CNS versus systemic) and the type (for example, infectious versus sterile) of the initial precipitating event (Glass et al., 2010). The BBB represents a key regulatory element of the communication between intrinsic brain cells and peripheral immunocompetent cells. As noted above, an inflammatory response in the CNS can be induced in the absence of infection. Brain inflammation has been reported following ischaemic stroke or traumatic brain injury (TBI), and during chronic neurodegenerative diseases. In all these conditions, pronounced activation of microglia and astrocytes takes place in brain regions affected by the specific disease, and these cells act as major sources of inflammatory mediators. Recruitment of peripheral immune cells might also occur (Nguyen et al., 2002; Glass et al., 2010). The activation of innate immunity and the transition to adaptive immunity are mediated by a large variety of inflammatory mediators, among which cytokines, polypeptides that act as soluble mediators of inflammation, have a pivotal role (Akira et al., 2001; Nguyen et al., 2002).
These molecules include interleukins (ILs), interferons (IFNs), tumour necrosis factors (TNFs) and growth factors (for example, transforming growth factor (TGF)b). Cytokines are released by immunocompetent and endothelial cells, as well as by glia and neurons in the CNS, thereby enabling communication between effector and target cells during an immune challenge or tissue injury. Following their release, cytokines interact with one or more cognate receptors. The most extensively studied prototypical inflammatory cytokines in the CNS are IL1b, TNF and IL6 (Allan and Rothwell, 2001; Bartfai et al., 2007). Cytokine activity can be regulated at multiple levels, including gene transcription, cleavage of cytokine precursors (for example, proIL1b, proTNF) by specific proteolytic enzymes, and cellular release, as well as through receptor signalling (discussed below). All cell types in the brain seem capable of expressing cytokines and their receptors, with low basal expression of these molecules being rapidly upregulated following CNS insults. Chemokines comprise a specific class of cytokines that act as chemoattractants to guide the migration of leukocytes from blood through the endothelial barrier into sites of infection or injury (Wilson et al., 2010). These cytokines also regulate microglial motility and neural stem cell migration, provide axon guidance during brain development, and promote angiogenesis, neurogenesis and synaptogenesis (Szekanecz and Koch, 2001; Semple et al., 2010). The release of chemokines is often stimulated by proinflammatory cytokines such as IL1b.
Several mechanisms have been identified that attenuate the inflammatory response, indicating the importance of such strict control for homeostasis and prevention of injury. Regulatory mechanisms include production of proteins that compete with cytokines to bind their receptors, such as IL1 receptor antagonist protein (IL1ra), and decoy receptors that bind cytokines and chemokines but are incapable of signalling, thereby acting as molecular traps to prevent such ligands from interacting with biologically active receptors (Mantovani et al., 2001; Dinarello, 2009). Proteins that inhibit cytokineinduced signal transduction (for example, suppressor of cytokine signalling proteins) or transcription (for example, Nurr1CoREST or activity transcription factor 3), as well as an array of soluble mediators with antiinflammatory activities (such as IL10 and TGFb), are produced concomitantly with proinflammatory molecules to resolve inflammation (Blobe et al., 2000; Khuu et al., 2007; Baker et al., 2009). For example, glucocorticoids, via activation of glucocorticoid receptors and, consequently, downregulation of nuclear factorkB (NFkB) and activator protein 1 activity, inhibit innate immune responses and, hence, act as an endogenous antiinflammatory feedback system. Proinflammatory cytokines are powerful enhancers of glucocorticoid levels in adrenal glands via corticotropinreleasing hormone and adrenocorticotropic hormone (ACTH). Glucocorticoids also elicit immunosuppressive effects through inhibition of leukocyte extravasation from the vasculature, and through regulation of T helper cell differentiation (Sapolsky et al., 1987; Elenkov et al., 1999). The CNS can also negatively regulate the inflammatory response in a reflexive manner, using the efferent activity of the vagus nerve to inhibit release of proinflammatory molecules from tissue macrophages (Vezzani et al., 2000a, 2011; Tracey, 2002).
Do seizures cause inflammation?
In adult rats and mice, induction of recurrent short seizures or single prolonged seizures (status epilepticus; defined as a seizure lasting >30 min) by chemoconvulsants or electrical stimulation triggers rapid induction of inflammatory mediators in brain regions of seizure activity onset and propagation (Vezzani et al., 2000a, 2011; Crespel et al., 2002). Immunohistochemical studies on rodent brains after induction of status epilepticus demonstrated subsequent waves of inflammation during the epileptogenic process (that is, the process underlying the onset and chronic recurrence of spontaneous seizures after an initial precipitating event), involving various cell populations. Findings from these and other studies show that proinflammatory cytokines (IL1b, TNF and IL6) are first expressed in activated microglia and astrocytes, and cytokine receptor expression is upregulated in microglia, astrocytes and neurons (Vezzani and Granata, 2005). These initial events are followed by the induction of cyclooxygenase2 (COX2) and, hence, prostaglandins, and upregulation of components of the complement system in microglia, astrocytes and neurons (Yoshikawa et al., 2006; Aronica et al., 2007; Kulkarni and Dhir, 2009; Xu et al., 2009).
In addition to the molecules mentioned above, chemokines and their receptors are produced – predominantly in neurons and in activated astrocytes – days to weeks after status epilepticus (Wu et al., 2008; Xu et al., 2009; Fabene et al., 2010). An ensuing wave of inflammation is induced in brain endothelial cells by seizures, and includes upregulation of IL1b and its receptor IL1R1, the complement system, and adhesion molecules (Pselectin, Eselectin, intercellular adhesion molecule 1 (ICAM) and vascular cell adhesion molecule 1) (Vezzani and Granata, 2005; Aronica et al., 2007; Fabene et al., 2008; Vezzani et al., 2011). The presumed cascade of events leading to this vascular inflammation involves seizureinduced activation of perivascular glia, which produce and release cytokines and prostaglandins. Importantly, no peripheral immune cells or bloodderived inflammatory molecules are required for vascular inflammation, as such events have been replicated in vitro in isolated guinea pig brain undergoing seizure activity (Vezzani and Granata, 2005; Vezzani et al., 2011).
The presence of inflammation originating from the brain might promote the recruitment of peripheral inflammatory cells. Indeed, chemokines expressed by neurons and glia and in the cerebrovasculature following seizures might direct blood leukocytes into the brain, which would be consistent with the reported emergence of granulocytes during epileptogenesis, and sparse T lymphocytes in chronic epileptic tissue from TLE models and humans (Ravizza et al., 2008). As in human epileptic brain specimens, brain tissue from rodents with experimental chronic TLE contains both activated astrocytes and microglia expressing inflammatory mediators (Crespel et al., 2002; Dube et al., 2007; Ravizza et al., 2008). Evidence for brain vessel inflammation associated with BBB breakdown is also prevalent (Fabene et al., 2008). A recent veterinary study evaluated the relationship of microglial activation to seizureinduced neuronal death in the cerebral cortex of Shetland sheepdogs with familial epilepsy (Sakurai et al., 2013). Cadavers of ten Shetland sheepdogs from the same family (six dogs with seizures and four dogs without seizures) and four agematched unrelated Shetland sheepdogs were evaluated. Samples of brain tissues were collected after euthanasia and sectioned for H&E staining and immunohistochemical analysis. Evidence of seizureinduced neuronal death was detected exclusively in samples of cerebral cortical tissue from the dogs with familial epilepsy in which seizures had been observed. The seizureinduced neuronal death was restricted to tissues from the cingulate cortex and sulci surrounding the cerebral cortex. In almost the same locations as where seizureinduced neuronal death was identified, microvessels appeared longer and more tortuous and the number of microvessels was greater than in the dogs without seizures and control dogs. Immunohistochemical results for neurons and glial cells (astrocytes and microglia) were positive for vascular endothelial growth factor, and microglia positive for ionized calciumbinding adapter molecule 1 were activated
(i.e. had swollen cell bodies and long processes) in almost all the same locations as where seizureinduced neuronal death was detected. Doublelabel immunofluorescence techniques revealed that the activated microglia had positive results for TNFa, IL6 and vascular endothelial growth factor receptor 1. These findings were not observed in the cerebrum of dogs without seizures, whether the dogs were from the same family as those with epilepsy or were unrelated to them. The suggested conclusion of this study was that microglial activation induced by vascular endothelial growth factor and associated proinflammatory cytokine production may accelerate seizureinduced neuronal death in dogs with epilepsy (Sakurai et al., 2013).
The findings discussed above show that brain inflammation induced by status epilepticus develops further during epileptogenesis and demonstrate that this phenomenon persists in chronic epileptic tissue, thereby supporting the idea that inflammation might be intrinsic to, and perhaps a biomarker of, the epileptogenic process (Dube et al., 2007).
Does inflammation cause seizures?
Although the functions of many inflammatory mediators remain unresolved, clear evidence exists for an active role for IL1b, TNF, IL6, prostaglandin E2 (PGE2) and the complement cascade in seizure generation and exacerbation (Xiong et al., 2003). Seizure activity leads to the production of inflammatory molecules that, in turn, affect seizure severity and recurrence, and this action takes place through mechanisms distinct from the transcriptional events traditionally activated during systemic inflammation. Cerebrospinal fluid studies in children and animal models have implicated the release of endogenous cytokines, especially IL1b, in the generation of febrile seizures and, possibly, in the development of epilepsy after febrile seizures (Haspolat et al., 2002; Virta et al., 2002; Dube et al., 2005; Heida and Pittman, 2005; Vezzani et al., 2013).
A positive feedback pathway has been identified in rat models between seizure activity and the presence of inflammation (Vezzani et al., 2011). However, the role of inflammation in epilepsy in veterinary medicine has really only been described clinically in cats with hippocampal necrosis (Fatzer et al., 2000). Hippocampal lesions of 38 cats with seizures have been described and seemed to reflect different stages of disease consisting of acute neuronal degeneration to complete malacia, affecting mainly the layer of the large pyramidal cells but sometimes also the neurons of the dentate gyrus and the piriform lobe. The clinical, neuropathologic and epidemiologic findings suggest that the seizures in these cats were triggered by primary structural brain damage, perhaps resulting from excitotoxicity, but secondary inflammation cannot be ruled out in these cases.
Does inflammation cause cell loss?
Available studies suggest that seizurerelated or injuryrelated inflammation might contribute to cell loss and synaptic reorganization, which are important mediators of the development of hyperexcitable circuits that lead to epilepsy after insults such as status epilepticus or TBI in the adult rodent brain (Bartfai and Schultzberg, 1993; Buckmaster and Dudek, 1997; Pitkanen and Sutula, 2002). Inflammation is induced rapidly following such insults, preceding neurodegeneration in lesional models of seizures (Rizzi et al., 2003; Ravizza and Vezzani, 2006). This finding is consistent with the idea that inflammation augments cell death, which is further supported by data from studies involving injection of inflammatory mediators together with excitotoxic stimuli (Allan et al., 2005). Activation of microglia and astrocytes and production of cytokines and PGE2 can occur in seizure models where cell loss is not detected in immature or adult rodents (Vezzani et al., 1999, 2000a; Rizzi et al., 2003; Kovacs et al., 2006; Dube et al., 2010). Such observations suggest that rather than being a consequence of cell loss, seizureinduced brain inflammation can contribute to cell death (Vezzani and Baram, 2007). Additional interactions between inflammation and cell death in the context of epilepsy have been observed. Brain injury, such as TBI, causes tissue inflammation that seems to contribute to both cell death and longterm hyperexcitability (Clausen et al., 2009; Longhi et al., 2009). In the context of CNS injury (for example, in chronic neurodegenerative diseases or acute stroke), inflammation can have a neuroprotective role (Liesz et al., 2009; Schwartz and Shechter, 2010). Indeed, whether microglia, macrophages and/or T cells are destructive or neuroprotective seems to depend on their activation status, which is orchestrated by the specific inflammatory environment (Rothwell, 1989; Schwartz and Shechter, 2010). This balance, together with the specific brain regions in which inflammation develops, might account for the relatively low incidence of seizures in other neurological disorders associated with brain inflammation (Vezzani et al., 2013).
mechanistic insights
Several established and novel mechanisms could mediate the effects of inflammatory mediators on neuronal excitability and epilepsy. Some of these mechanisms could be involved in the precipitation and recurrence of seizures, while others are implicated in the development of epileptogenesis (Vezzani and Baram, 2007). These mechanisms constitute potential molecular targets for drug design, and are briefly summarized here. As discussed above, IL1b and HMGB1 activate convergent signalling cascade through binding to IL1R1 and TLR4, respectively (Akira et al., 2001; Perkins, 2007; Hoebe and Beutler, 2008). The downstream pathways activated by these ligands converge with the TNF pathways at the transcription factor NFkB, which regulates the synthesis of chemokines, cytokines, enzymes (for example, COX2) and receptors (for example, TLRs, IL1R1, and TNF p55 and p75 receptors) (Gilmore, 2006). This transcriptional pathway modulates the expression of genes involved in neurogenesis, cell death and survival, and in synaptic molecular reorganization and plasticity (processes that occur concomitantly with epileptogenesis in experimental models) (Buckmaster and Dudek, 1997; Pitkanen and Lukasiuk, 2009).
immune and anti-inflammatory therapies
If immune mechanisms and inflammation do indeed have a role in the generation of seizures, immunemodulating and antiinflammatory therapies might be effective treatments for some or all forms of epilepsy. Therapies such as ACTH, corticosteroids, plasmapheresis and intravenous immunoglobulin (IVIg) have been employed to treat seizures and/or epilepsy, with varying success. These therapies have all been employed in human patients with presumed autoimmune limbic encephalitis, where early and aggressive treatment often seems to be useful (Vincent et al., 2010).
The presumed mechanism of action of the therapeutic agents listed above is suppression of inflammation; however, other modes of action might also be involved, including direct effects on brain excitability, and suppression of endogenous proconvulsant brain agents (Baram and Hatalski, 1998; Joels and Baram, 2009).
The use of steroids in various forms is common for more severe, treatmentresistant forms of childhood epilepsy. ACTH, steroids and IVIg have all been employed to treat AEMunresponsive paediatric epilepsies, difficult focal (partial) epilepsies and myoclonic epilepsies (You et al., 2008). Unfortunately, determination of whether patients received benefit from these treatments is problematic, since most of these epilepsies are extremely heterogeneous in aetiology and severity, and exhibit notoriously variable courses. In addition, most of the clinical studies are retrospective case series, with occasional prospective case series that lack controls (Mikati et al., 2002; Verhelst et al., 2005).
Followup duration in these case series was also often variable. A recent review of investigations of IVIg in intractable childhood epilepsy found no randomized or controlled studies and, in fact, only two case series employed statistics in assessing outcome (Mikati et al., 2010). One series showed a statistically significant reduction in seizures with IVIg treatments, while the other revealed an insignificant trend with such therapy (Mikati et al., 2010). However, a Cochrane Collaboration review on the use of ACTH for other childhood epilepsies, published in 2007, found only a single randomized controlled trial, which only included five patients (Gayatri et al., 2007). The authors of this review concluded that, at present, no evidence exists to support either the safety or the efficacy of ACTH for general paediatric epilepsies (Gayatri et al., 2007).
Disorders of Neuronal migration and Seizures
The major developmental disorders noted in humans giving rise to epilepsy are disorders of neuronal migration that may have genetic or intrauterine causes (Engelborghs et al., 2000). Abnormal patterns of neuronal migration lead to various forms of agyria or pachygyria whereas lesser degrees of failure of neuronal migration induce neuronal heterotopia in the subcortical white matter. Experimental data suggest that cortical malformations can both form epileptogenic foci and alter brain development such that diffuse hyperexcitability of the cortical network occurs (ChevassusauLouis et al., 1999). Other studies revealed increases in postsynaptic glutamate receptors and decreases in GABAA receptors in microgyric cortex, which could promote epileptogenesis (Jacobs et al., 1999).
Periventricular heterotopia is a human Xlinked dominant disorder of cerebral cortical development. Mutations in the filamin 1 gene prevent migration of cerebral cortical neurons causing periventricular heterotopia (Fox et al., 1998). Affected females present with epilepsy whereas affected males die embryonically.
Lissencephaly is a brain malformation characterized by a paucity of gyral formation and a thickening of the cerebral cortex. It is presumed to occur secondary to incomplete migration of immature neurons to the cortical plate during fetal development (Saito et al., 2002). Lissencephaly is considered to be the most severe type of neuronal migration disorder compatible with survival. In humans, it is presumed to result from an arrest of neuronal migration at approximately 3 to 4 months (Dobyns et al., 1993). Once they exit the cell cycle in the periventricular proliferative zone, immature neurons must migrate to the cortical plate along radial glial fibres (Rakic, 1988). The six layers of the cerebral cortex are formed in an ‘inside out’ pattern, with early migrating neurons forming the deep layer and later migrating neurons passing their migratory predecessors to form the superficial layers. Interruption at any stage of the process of neuronal migration may result in the arrest of neurons in an intermediate position between the periventricular zone and the cortex (Saito et al., 2002). Such an interruption may be due to a genetic lack of appropriate molecular cues, or secondary to nongenetic influences such as in utero infection or ischaemia. Secondary influences are a more common mechanism for the related cortical malformation, polymicrogyria.
In humans, mutations of two genes, LIS1 (located on 17p13.3) and DCX (located on Xq22.3), have been found to account for the majority of cases (Pilz et al., 1998). Both of these genes have been shown to have roles in neuronal migration by their interactions with the neuron microtubule network (Gleeson et al., 1999a; Sapir et al., 1999). Xlinked lissencephaly and double cortex syndrome is a disorder of neuronal migration documented in humans. Double cortex or subcortical band heterotopias often occur in females whereas more severe lissencephaly is found in affected males. A causal mutation in a gene called doublecortin has been identified (Gleeson et al., 1998). It was suggested that doublecortin acts as an intracellular signalling molecule critical for the migration of developing neurons (Allen and Walsh, 1999; Gleeson et al., 1999b). Lissencephaly has been documented in Lhasa apsos with histopathology indicating the condition to be very similar to that seen in people (Greene et al., 1976; Saito et al., 2002). This condition has also been documented in a mixed breed dog and together with either cerebellar hypoplasia in two wirehaired fox terriers and three Irish setters, with cyclopia in one German shepherdmixed breed dog, or with microencephaly in the Korat breed of cat (Saito et al., 2002; Lee et al., 2011).
Although these disorders are relatively rare, studying the underlying pathophysiological mechanisms may shed light on the pathophysiology of more common epileptic syndromes.
How Do Seizures Stop?
Most seizures are selflimited, lasting no more than a few minutes. The persistence of a seizure lasting longer than several minutes is usually a cause for alarm as physiological mechanisms terminating the seizure may have failed. Why seizures typically do not continue indefinitely, and how intrinsic anticonvulsant mechanisms in the brain lead to seizure termination, are questions that potentially offer new avenues for developing novel treatments for epilepsy, as well as offering insights into brain autoregulatory mechanisms.
mechanisms acting at the level of single neurons
Within a single neuron, prolonged depolarizations with sustained actionpotential firing may be triggered by a brief depolarizing pulse, as in the paroxysmal depolarizing shift, or may be the result of sustained excitatory synaptic input from neighbouring neurons engaged in seizure activity (Ayala, 1983). Intrinsic mechanisms of seizure termination active in a single neuron, discussed below, include: the potassium currents activated by calcium and sodium entry; the loss of ionic gradients, particularly of potassium, leading first to depolarization with increased firing, followed by depolarization blockade of membrane firing and cessation of firing; and possibly the depletion of energy substrates locally, with the decline in adenosine triphosphate (ATP), resulting in cessation of neuronal firing.
Intracellular ion-activated potassium currents
The membrane after hyperpolarization that follows bursts of action potential discharge is the result, at least in part, of potassium currents activated by the entry of calcium and sodium. Increased calcium entry during the paroxysmal depolarizing shift, or as a result of the action of glutamate at the postsynaptic membrane, activates a calciumdependent membrane potassium conductance that allows potassium efflux, membrane hyperpolarization and cessation of firing (Alger and Nicoll, 1980; Timofeev et al., 2004). Like calcium, sodium entry may also activate a sodiumdependent potassium current that reduces neuronal excitability by hyperpolarizing the membrane and increasing shunt conductance (Schwindt et al., 1989).
Transmembrane ion gradients
The effect of extracellular potassium is multifaceted. Sustained potassium efflux increases extracellular potassium concentration, depolarizing the membrane and moving the intracellular voltage toward the threshold for sodium action potential firing. As extracellular potassium continues to accumulate, there is membrane depolarization and action potential firing increases. With further accumulation, the membrane potential becomes more depolarized than the firing threshold for sodiumaction potentials, sodium channels inactivate, and neuronal firing ceases. In vitro experiments by Bikson et al. (2003) illustrate these effects of extracellular potassium accumulation. Electrographic seizurelike activity triggered in hippocampal slices by exposure to lowcalcium artificial cerebrospinal fluid (aCSF) manifested as recurrent periods of population firing followed by periods of electrographic silence lasting 12–18 s. The termination of each electrographic discharge by a period of electrographic silence resulted from transient increases in extracellular potassium to plateaus of approximately 12 mM. The depolarized state was maintained by the elevation of extracellular potassium and by the presence of persistent sodium channels that did not inactivate. Depolarization blockadeterminating seizurelike discharges have also been observed in neocortical slices in which GABAergic inhibition is partially blocked by picrotoxin (Pinto et al., 2005). Focal or localized increases in potassium may also trigger additional potassium release beyond the initial region of potassium accumulation. Shifts in extracellular potential, and oscillations seen at the end of hippocampal afterdischarges, have been attributed to a rapid rise in extracellular potassium that triggers waves of astrocyte depolarization and a propagating rise in potassium that terminates neuronal firing (Bragin et al., 1997). In addition to its direct depolarizing effects, increased extracellular potassium may also indirectly result in membrane depolarization through the action of the potassium–chloride cotransporter KCC2. The rise in extracellular potassium can increase intracellular chloride, shifting the chloride reversal potential toward membrane depolarization. In the setting of increased intracellular chloride, the action of GABA to open chloride channels could enhance membrane depolarization to the point of becoming refractory to further firing of action potentials (Jin et al., 2005; Galanopoulou, 2007).
Extracellular calcium levels also change markedly during paroxysmal neuronal firing and may affect the efficiency of neurontoneuron spread of activity. Focal seizure activity results in a decline in extracellular calcium activity of approximately 50% (Heinemann et al., 1977). This decline may inhibit synaptic transmission because synaptic vesicle fusion and neurotransmitter release are dependent on entry of extracellular calcium (King et al., 2001; Cohen and Fields, 2004). Decline in extracellular calcium also potentially affects gap junction function as hemichannel opening increases in low calcium (Thimm et al., 2005).
Energy failure
Sustained neuronal activation also markedly increases energy, namely ATP, utilization to restore ion gradients across the membrane. In some neurons, the presence of an ATPgated potassium channel (KATP) reduces neuronal activity when ATP levels decline intracellularly (Yamada et al., 2001). When the ATP level falls because energy utilization outpaces energy production, potassium channels open and produce membrane hyperpolarization. Indeed, knockout mice lacking functioning KATP channels experience a myoclonic seizure on average 8.9 ± 1.1 s following onset of hypoxia, followed by generalized convulsions and death. A similar hypoxic challenge, however, does not trigger seizures in wildtype mice, indicating that KATP channels in vivo resist membrane depolarization during energy failure. Reduced levels of energy metabolites, such as glucose, may also affect seizure duration. In vitro recordings show that decreasing extracellular glucose terminates electrographic seizurelike activity in the low magnesium hippocampal slice (Kirchner et al., 2006). The effect of hypoglycaemia on seizurelike discharges in vitro was statistically significant, but not immediate. Fifty per cent fewer seizurelike discharges occurred in the 24min period following application of low glucose artificial cerebrospinal fluid compared to the frequency of discharges in the 30 min prior to application. Low glucose also reduced the amplitude of the seizurelike discharge by 25%. These effects on the frequency and amplitude of seizurelike discharges were reversed by restoration of normal glucose levels.
mechanisms acting on a local network of neurons
While seizure initiation is driven at least in part by the burstfiring properties of the individual neurons, the evolution and spread of the seizures also requires amplification and synchronization among neurons within susceptible networks. Seizure amplification occurs through the action of recurrent excitatory collaterals that form feedback loops, returning excitatory synaptic activity to the neurons within the seizure onset zone (Rutecki et al., 1989; Coulter and DeLorenzo, 1999). Seizure spread depends on the propagation and synchronization of the seizure discharge across synapses that separate neurons in the seizure onset zone from ‘normal’ neurons synaptically connected to the seizure onset zone (MacVicar and Dudek, 1980; Miles and Wong, 1983).
Glutamate depletion
Decrease in synaptic efficacy results in milder postsynaptic excitation, and consequently diminished amplification and spread of the seizure discharge. One mechanism limiting synaptic transmission during a sustained seizure discharge is the depletion of synaptic vesicles containing neurotransmitter. Staley et al. (1998) investigated the effects of synaptic depletion in vitro using a model of CA3 electrographic seizure discharges produced by hyperkalaemia. CA3 discharges consist of recurrent neuronal depolarizations with bursts of actionpotential firing separated by period of electrographic silence. Staley et al. found that the duration of the seizure burst was proportional to the duration of the silent period preceding the burst, consistent with the hypothesis that the seizure burst duration depended on the renewed availability of immediately releasable glutamate. If glutamatecontaining synaptic vesicles are replaced at a steady rate, longer interburst periods allow a greater resupply of immediately releasable glutamate, and an increased duration of the subsequent electrographic seizure discharge. Interburst intervals of 2–3 s or longer were necessary to achieve the longest burst durations (up to 420 ms). Thus, as the seizure discharge develops, it consumes the supply of readily releasable glutamate needed to sustain the seizure, potentially acting as a governor on excitatory drive. As the glutamate reservoir is replenished continuously, however, additional control mechanisms are necessary to prevent reinitiation of seizure activity.
The intra- and extracellular environments
Prolonged neuronal activity during seizure discharges may also have the effect of increasing CO2 or increasing the byproducts of anaerobic metabolism, and produce extracellular acidosis or intracellular acidosis associated with extracellular alkalinosis (Chesler and Kaila, 1992). Glial cells may also contribute to acidification of the extracellular space in response to increases in the extracellular potassium concentration (Chesler and Kraig, 1987). In the hippocampal slice in vitro, acidification of the extracellular space to pH 6.7 terminated seizurelike burst firing facilitated by lowmagnesium in the artificial CSF. The attenuation of epileptiform activity began within minutes of lowering pH (Velisek et al., 1994; Velisek, 1998). The mechanisms of action – at least in part – included decreased NMDA receptor function and loss of synaptic longterm potentiation (LTP). A milder reduction of pH to 7.1 also produced milder synaptic impairment with continued loss of LTP (Velisek, 1998). Inhibition of carbonic anhydrase, which alters extracellular pH, has some anticonvulsant benefit. In humans, the carbonic anhydrase inhibitor acetozolamide has a mild anticonvulsant effect (Thiry et al., 2007). Knockout mice deficient in carbonic anhydrase are severely acidotic and are resistant to seizures produced by flurothyl gas compared to wildtype mice (Velisek et al., 1993). Intracellular acidification may also contribute to termination of seizure discharges. Spontaneous interictal spiking following focal application of bicuculline in the piriform cortex in an in vitro whole brain preparation was associated with periodic abrupt alkanization of the extracellular space followed by a slow return to baseline pH (de Curtis et al., 1998). These observations were interpreted as evidence of intracellular acidification. Application of ammonium chloride in the perfusing medium to prevent intracellular acidification increased neuronal excitability and resulted in afterdischarges following each spike, and in seizurelike discharges. The investigators hypothesized that the intracellular acidification reduced excitability by reducing gapjunction function. Application of octanol, a nonspecific gap junction blocker, abolished spontaneous interictal spiking (de Curtis et al., 1998).
Glial buffering of glutamate
Glial uptake of perisynaptic glutamate is the major mechanism forestalling accumulation of glutamate at the synapse (Benarroch, 2005). Astrocytes have an equally important role in the regulation of extracellular potassium. Astrocytic buffering of potassium maintains extracellular levels below a ceiling of 12 mM (Benarroch, 2005). In some cases, such as the epileptic brain, glia may also release glutamate, thereby prolonging postsynaptic excitation. Tian et al. (2005) recently showed that glial release of glutamate contributed to the maintenance of the paroxysmal depolarizing shift that is the hallmark of ‘epileptic’ neurons. Failure of glia to buffer extracellular glutamate, let alone glutamate release from glia, can be expected to result in prolonged excitatory drive and seizure maintenance.
Increased GABA-ergic inhibition
A basic mechanism to control focal seizure activity is GABAergic synaptic inhibition mediated by local interneurons. Seizure discharges within the seizure onset zone produce recurrent inhibition within the seizure initiation zone, thus reducing excitatory output (Kostopoulos et al., 1983; Dorn and Witte, 1995). Early investigations of the spike and wave components of ‘spikewave’ discharges showed that the spike component is associated with a burst of rapid actionpotential firing, while the wave component is associated with a pause in actionpotential firing (Dichter and Spencer, 1969). The pause in neuronal firing results from synaptic inhibition produced by local inhibitory interneurons activated by the volley of excitatory activity comprising the ‘spike’ component, an example of feedback inhibition. Feedforward inhibition is a fundamental feature of cortical processing (Swadlow, 2003). Feedforward inhibition may also play an important role; an interneuron activated by a principal cell sends inhibitory signals to principal cells outside the focus, inhibiting the propagation of the seizure (Trevelyan et al., 2007). Recent evidence indicates that a principal cell axon may synapse on the presynaptic terminal of an inhibitory interneuron, bypassing somatic activation of the interneuron altogether by causing transmitter release directly from the inhibitory synaptic terminal (Connors and Cruikshank, 2007).
Synaptic inhibition is mediated by the presynaptic release of the neurotransmitter GABA, which acts on the postsynaptic neuron via receptors located on the soma, dendrites, or presynaptic terminals. GABA receptors are present in two major varieties, GABAA and GABAB. GABAB receptors are metabotropic acting through Gprotein second messengers. The preand postsynaptic distribution of GABAB receptors, along with mixed evidence of anti and proconvulsant effects of GABAB activation, makes it difficult to determine their role in seizure termination (Chen et al., 2004). GABAA receptors are chlorideconducting membrane channels that open rapidly in response to GABA. Desensitization of GABAA receptors during status epilepticus likely contributes to the failure of seizure termination (Chen et al., 2007). Desensitization of GABAA receptors is also the basis of the loss of efficacy of benzodiazepine medications used to treat status epilepticus. Multiple mechanisms appear to contribute to GABAA receptor desensitization. Increased internalization of GABAA receptors during status epilepticus reduces the effect of GABAergic stimulation (Goodkin et al., 2007). Changes in subunit composition may also contribute to GABAA receptor desensitization, although this process acts over many minutes to hours, and appears to affect longterm neuronal excitability and epileptogenesis rather than seizure termination. Nonsynaptic GABAA receptors, in contrast, do not desensitize and instead are capable of tonic inhibition, which produces longlasting changes in neuronal reactivity. These tonic GABA receptors typically contain particular subunits – delta and possibly gamma – that alter the properties of the receptors (Richerson, 2004). Tonic receptors are activated by micromolar levels of the extrasynaptic GABA, which arrives either by diffusion from a synapse before reuptake, or by release into the extracellular space via nonsynaptic mechanisms. Tonic GABA receptors may play an important role in epilepsy. On the one hand, reduction of tonic GABA currents (as produced experimentally by a mutation in the deltasubunit of tonic GABA receptors) is associated with generalized epilepsy. On the other hand, progesteronederived neurosteroids enhance tonic GABA currents, and may play a role in preventing seizure genesis, and potentially in terminating ongoing seizures (Stell et al., 2003). It is also clear that the contribution of extrasynaptic GABAA receptors changes during maturation, and may contribute to changes in seizure susceptibility during development.
references
Aarli, J.A. (2000) Epilepsy and the immune system. Archives of Neurology 57, 1689–1692.
Akira, S., Takeda, K. and Kaisho, T. (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunology 2, 675–680.
Alger, B.E. and Nicoll, R.A. (1980) Epileptiform burst after hyperolarization: calcium-dependent potassium potential in hippocampal CA1 pyramidal cells. Science 210, 1122–1124.
Allan, S.M. and Rothwell, N.J. (2001) Cytokines and acute neurodegeneration. Nature Reviews Neuroscience 2, 734–744.
Allan, S.M., Tyrell, P.J. and Rothwell, N.J. (2005) Interleukin-1 and neuronal injury. Nature Reviews Immunology 5, 629–640.
Allen, K.M. and Walsh, C.A. (1999) Genes that regulate neuronal migration in the cerebral cortex. Epilepsy Research 36, 143–154.
Angehagen, M., Ben-Menachem, E., Ronnback, L. and Hansson, E. (2003a) Novel mechanisms of action of three antiepileptic drugs, vigabatrin, tiagabine, and topiramate. Neurochemical Research 28, 333–340.
Angehagen, M., Ben-Menachem, E., Ronnback, L. and Hansson, E. (2003b) Topiramate protects against glutamate- and kainate-induced neurotoxicity in primary neuronal-astroglial cultures. Epilepsy Research 54, 63–71.
Aronica, E., Boer, K., Van Vliet, E.A., Redeker, S., Baayen, J.C., Spliet, W.G., Van Rijen, P.C., Troost, D., Da Silva, F.H., Wadman, W.J. and Gorter, J.A. (2007) Complement activation in experimental and human temporal lobe epilepsy. Neurobiology of Disease 26, 497–511.
Ayala, G.F. (1983) The paroxysmal depolarizing shift. Progress in Clinical and Biological Research 124, 15–21.
Baker, B.J., Akhtar, L.N. and Benveniste, E.N. (2009) SOCS1 and SOCS3 in the control of CNS immunity. Trends in Immunology 30, 392–400.
Balosso, S., Ravizza, T., Perego, C., Peschon, J., Campbell, I.L., De Simoni, M.G. and Vezzani, A. (2005) Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Annals of Neurology 57, 804–812.
Banks, W.A. and Erickson, M.A. (2010) The blood-brain barrier and immune function and dysfunction. Neurobiology of Disease 37, 26–32.
Baram, T.Z. and Hatalski, C.G. (1998) Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends in Neuroscience 21, 471–476.
Bartfai, T. and Schultzberg, M. (1993) Cytokines in neuronal cell types. Neurochemistry International 22, 435–444.
Bartfai, T., Sanchez-Alavez, M., Andell-Jonsson, S., Schultzberg, M., Vezzani, A., Danielsson, E. and Conti, B. (2007) Interleukin-1 system in CNS stress: seizures, fever, and neurotrauma. Annals of the New York Academy of Science 1113, 173–177.
Benarroch, E.E. (2005) Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clinic Proceedings 80, 1326–1338.
Bengzon, J., Okabe, S., Lindvall, O. and Mckay, R.D. (1999) Suppression of epileptogenesis by modification of N-methyl-D-aspartate receptor subunit composition. European Journal of Neuroscience 11, 916–922.
Bien, C.G., Urbach, H., Schramm, J., Soeder, B.M., Becker, A.J., Voltz, R., Vincent, A. and Elger, C.E. (2007) Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology 69, 1236–1244.
Bikson, M., Hahn, P.J., Fox, J.E. and Jefferys, J.G. (2003) Depolarization block of neurons during maintenance of electrographic seizures. Journal of Neurophysiology 90, 2402–2408.
Blobe, G.C., Schiemann, W.P. and Lodish, H.F. (2000) Role of transforming growth factor beta in human disease. New England Journal of Medicine 342, 1350–1358.
Bordey, A. and Sontheimer, H. (1998) Properties of human glial cells associated with epileptic seizure foci. Epilepsy Research 32, 286–303.
Bragin, A., Penttonen, M. and Buzsaki, G. (1997) Termination of epileptic afterdischarge in the hippocampus. Journal of Neuroscience 17, 2567–2579.
Buckmaster, P.S. and Dudek, F.E. (1997) Network properties of the dentate gyrus in epileptic rats with hilar neuron loss and granule cell axon reorganization. Journal of Neurophysiology 77, 2685–2696.
Campbell, I.L., Abraham, C.R., Masliah, E., Kemper, P., Inglis, J.D., Oldstone, M.B. and Mucke, L. (1993) Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proceedings of the National Academies of Science USA 90, 10061–10065.
Cavazos, J.E., Golarai, G. and Sutula, T.P. (1991) Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. Journal of Neuroscience 11, 2795–2803.
Chapman, A.G. (1998) Glutamate receptors in epilepsy. Progress in Brain Research 116, 371–383.
Chapman, A.G. (2000) Glutamate and epilepsy. Journal of Nutrition 130, 1043S–1045S.
Chapman, A.G., Elwes, R.D., Millan, M.H., Polkey, C.E. and Meldrum, B.S. (1996) Role of glutamate and aspartate in epileptogenesis; contribution of microdialysis studies in animal and man. Epilepsy Research Suppl. 12, 239–246.
Chen, J.W., Naylor, D.E. and Wasterlain, C.G. (2007) Advances in the pathophysiology of status epilepticus. Acta Neurologica Scandinavica 115, 7–15.
Chen, L., Chan, Y.S. and Yung, W.H. (2004) GABA-B receptor activation in the rat globus pallidus potently suppresses pentylenetetrazol-induced tonic seizures. Journal of Biomedical Science 11, 457–464.
Chesler, M. and Kaila, K. (1992) Modulation of pH by neuronal activity. Trends in Neuroscience 15, 396–402.
Chesler, M. and Kraig, R.P. (1987) Intracellular pH of astrocytes increases rapidly with cortical stimulation. American Journal of Physiology 253, R666–670.
Chevassus-Au-Louis, N., Baraban, S.C., Gaiarsa, J.L. and Ben-Ari, Y. (1999) Cortical malformations and epilepsy: new insights from animal models. Epilepsia 40, 811–821.
Choi, J., Nordli, D.R., Jr, Alden, T.D., Dipatri, A., Jr, Laux, L., Kelley, K., Rosenow, J., Schuele, S.U., Rajaram, V. and Koh, S. (2009) Cellular injury and neuroinflammation in children with chronic intractable epilepsy. Journal of Neuroinflammation 6, 38.
Clausen, F., Hanell, A., Bjork, M., Hillered, L., Mir, A.K., Gram, H. and Marklund, N. (2009) Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. European Journal of Neuroscience 30, 385–396.
Cohen, J.E. and Fields, R.D. (2004) Extracellular calcium depletion in synaptic transmission. Neuroscientist 10, 12–17.
Connors, B.W. and Cruikshank, S.J. (2007) Bypassing interneurons: inhibition in neocortex. Nature Neuroscience 10, 808–810.
Coulter, D.A. and DeLorenzo, R.J. (1999) Basic mechanisms of status epilepticus. Advances in Neurology 79, 725–733.
Crespel, A., Coubes, P., Rousset, M.C., Brana, C., Rougier, A., Rondouin, G., Bockaert, J., Baldy-Moulinier, M. and Lerner-Natoli, M. (2002) Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Research 952, 159–169.
D’Hooge, R., Raes, A., Lebrun, P., Diltoer, M., Van Bogaert, P.P., Manil, J., Colin, F. and De Deyn, P.P. (1996) N-methyl-D-aspartate receptor activation by guanidinosuccinate but not by methylguanidine: behavioural and electrophysiological evidence. Neuropharmacology 35, 433–440.
De Curtis, M., Manfridi, A. and Biella, G. (1998) Activity-dependent pH shifts and periodic recurrence of spontaneous interictal spikes in a model of focal epileptogenesis. Journal of Neuroscience 18, 7543–7551.
De Deyn, P.P. and Macdonald, R.L. (1990) Guanidino compounds that are increased in cerebrospinal fluid and brain of uremic patients inhibit GABA and glycine responses on mouse neurons in cell culture. Annals of Neurology 28, 627–633.
De Deyn, P.P., Marescau, B. and Macdonald, R.L. (1990) Epilepsy and the GABA-hypothesis a brief review and some examples. Acta Neurologica Belgica 90, 65–81.
Dichter, M.A. and Ayala, G.F. (1987) Cellular mechanisms of epilepsy: a status report. Science 237, 157–164.
Dichter, M. and Spencer, W.A. (1969) Penicillin-induced interictal discharges from the cat hippocampus. II. Mechanisms underlying origin and restriction. Journal of Neurophysiology 32, 663–687.
Dinarello, C.A. (2009) Immunological and inflammatory functions of the interleukin-1 family. Annual Review of Immunology 27, 519–550.
Dobyns, W.B., Reiner, O., Carrozzo, R. and Ledbetter, D.H. (1993) Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. Journal of the American Medical Association 270, 2838–2842.
Doherty, J. and Dingledine, R. (2002) The roles of metabotropic glutamate receptors in seizures and epilepsy. Current Drug Targets: CNS and Neurological Disorders 1, 251–260.
Dorn, T. and Witte, O.W. (1995) Refractory periods following interictal spikes in acute experimentally induced epileptic foci. Electroencephalography and Clinical Neurophysiology 94, 80–85.
Dube, C., Vezzani, A., Behrens, M., Bartfai, T. and Baram, T.Z. (2005) Interleukin-1beta contributes to the generation of experimental febrile seizures. Annals of Neurology 57, 152–155.
Dube, C.M., Brewster, A.L., Richichi, C., Zha, Q. and Baram, T.Z. (2007) Fever, febrile seizures and epilepsy. Trends in Neuroscience 30, 490–496.
Dube, C.M., Ravizza, T., Hamamura, M., Zha, Q., Keebaugh, A., Fok, K., Andres, A.L., Nalcioglu, O., Obenaus, A., Vezzani, A. and Baram, T.Z. (2010) Epileptogenesis provoked by prolonged experimental febrile seizures: mechanisms and biomarkers. Journal of Neuroscience 30, 7484–7494.
Duffy, S. and MacVicar, B.A. (1999) Modulation of neuronal excitability by astrocytes. Advances in Neurology 79, 573–581.
Dzhala, V.I., Talos, D.M., Sdrulla, D.A., Brumback, A.C., Mathews, G.C., Benke, T.A., Delpire, E., Jensen, F.E. and Staley, K.J. (2005) NKCC1 transporter facilitates seizures in the developing brain. Nature Medicine 11, 1205–1213.
Elenkov, I.J., Webster, E.L., Torpy, D.J. and Chrousos, G.P. (1999) Stress, corticotropin-releasing hormone, glucocorticoids, and the immune/inflammatory response: acute and chronic effects. Annals of the New York Academy of Science 876, 1–11; discussion 11–13.
Engelborghs, S., Marien, P., Martin, J.J. and De Deyn, P.P. (1998a) Functional anatomy, vascularisation and pathology of the human thalamus. Acta Neurologia Belgica 98, 252–265.
Engelborghs, S., Pickut, B.A., D’Hooge, R., Wiechert, P., Haegele, K. and De Deyn, P.P. (1998b) Behavioral effects of vigabatrin correlated with whole brain gamma-aminobutyric acid metabolism in audiogenic sensitive rats. Arzneimittelforschung 48, 713–716.
Engelborghs, S., D’Hooge, R. and De Deyn, P.P. (2000) Pathophysiology of epilepsy. Acta Neurologica Belgica 100, 201–213.
Errante, L.D. and Petroff, O.A. (2003) Acute effects of gabapentin and pregabalin on rat forebrain cellular GABA, glutamate, and glutamine concentrations. Seizure 12, 300–306.
Fabene, P.F., Navarro Mora, G., Martinello, M., Rossi, B., Merigo, F., Ottoboni, L., Bach, S., Angiari, S., Benati, D., Chakir, A., Zanetti, L., Schio, F., Osculati, A., Marzola, P., Nicolato, E., Homeister, J.W., Xia, L., Lowe, J.B., Mcever, R.P., Osculati, F., Sbarbati, A., Butcher, E.C. and Constantin, G. (2008) A role for leukocyteendothelial adhesion mechanisms in epilepsy. Nature Medicine 14, 1377–1383.
Fabene, P.F., Bramanti, P. and Constantin, G. (2010) The emerging role for chemokines in epilepsy. Jounal of Neuroimmunology 224, 22–27.
Fatzer, R., Gandini, G., Jaggy, A., Doherr, M. and Vandevelde, M. (2000) Necrosis of hippocampus and piriform lobe in 38 domestic cats with seizures: a retrospective study on clinical and pathologic findings. Journal of Veterinary Internal Medicine 14, 100–104.
Fellin, T. and Haydon, P.G. (2005) Do astrocytes contribute to excitation underlying seizures? Trends in Molecular Medicine 11, 530–533.
Fisher, R.S. (1989) Animal models of the epilepsies. Brain Research - Brain Research Reviews 14, 245–278.
Fox, J.W., Lamperti, E.D., Eksioglu, Y.Z., Hong, S.E., Feng, Y., Graham, D.A., Scheffer, I.E., Dobyns, W.B., Hirsch, B.A., Radtke, R.A., Berkovic, S.F., Huttenlocher, P.R. and Walsh, C.A. (1998) Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325.
Fukuda, A. and Prince, D.A. (1992) Postnatal development of electrogenic sodium pump activity in rat hippocampal pyramidal neurons. Brain Research - Developmental Brain Research 65, 101–114.
Galanopoulou, A.S. (2007) Developmental patterns in the regulation of chloride homeostasis and GABA(A) receptor signaling by seizures. Epilepsia 48 (Suppl 5), 14–18.
Gayatri, N.A., Ferrie, C.D. and Cross, H. (2007) Corticosteroids including ACTH for childhood epilepsy other than epileptic spasms. Cochrane Database System Reviews, CD005222.
Gerber, K., Filakovszky, J., Halasz, P. and Bagdy, G. (1998) The 5-HT1A agonist 8-OH-DPAT increases the number of spike-wave discharges in a genetic rat model of absence epilepsy. Brain Research 807, 243–245.
Gilmore, T.D. (2006) Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25, 6680–6684.
Glass, C.K., Saijo, K., Winner, B., Marchetto, M.C. and Gage, F.H. (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934.
Gleeson, J.G., Allen, K.M., Fox, J.W., Lamperti, E.D., Berkovic, S., Scheffer, I., Cooper, E.C., Dobyns, W.B., Minnerath, S.R., Ross, M.E. and Walsh, C.A. (1998) Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72.
Gleeson, J.G., Lin, P.T., Flanagan, L.A. and Walsh, C.A. (1999a) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23, 257–271.
Gleeson, J.G., Minnerath, S.R., Fox, J.W., Allen, K.M., Luo, R.F., Hong, S.E., Berg, M.J., Kuzniecky, R., Reitnauer, P.J., Borgatti, R., Mira, A.P., Guerrini, R., Holmes, G.L., Rooney, C.M., Berkovic, S., Scheffer, I., Cooper, E.C., Ricci, S., Cusmai, R., Crawford, T.O., Leroy, R., Andermann, E., Wheless, J.W., Dobyns, W.B., Walsh, C.A. et al. (1999b) Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Annals of Neurology 45, 146–153.
Goddard, G.V. (1967) Development of epileptic seizures through brain stimulation at low intensity. Nature 214, 1020–1021.
Goodkin, H.P., Sun, C., Yeh, J.L., Mangan, P.S. and Kapur, J. (2007) GABA(A) receptor internalization during seizures. Epilepsia 48 (Suppl 5), 109–113.
Greene, C.E., Vandevelde, M. and Braund, K. (1976) Lissencephaly in two Lhasa Apso dogs. Journal of the American Veterinary Medical Association 169, 405–410.
Grisar, T., Guillaume, D. and Delgado-Escueta, A.V. (1992) Contribution of Na+,K(+)-ATPase to focal epilepsy: a brief review. Epilepsy Research 12, 141–149.
Haglund, M.M., Stahl, W.L., Kunkel, D.D. and Schwartzkroin, P.A. (1985) Developmental and regional differences in the localization of Na,K-ATPase activity in the rabbit hippocampus. Brain Research 343, 198–203.
Hara, M., Sasa, M., Kawabata, A., Serikawa, T., Yamada, T., Yamada, J. and Takaori, S. (1993) Decreased dopa-mine and increased norepinephrine levels in the spontaneously epileptic rat, a double mutant rat. Epilepsia 34, 433–440.
Haspolat, S., Mihci, E., Coskun, M., Gumuslu, S., Ozben, T. and Yegin, O. (2002) Interleukin-1beta, tumor necrosis factor-alpha, and nitrite levels in febrile seizures. Journal of Child Neurology 17, 749–751.
Heida, J.G. and Pittman, Q.J. (2005) Causal links between brain cytokines and experimental febrile convulsions in the rat. Epilepsia 46, 1906–1913.
Heinemann, U., Lux, H.D. and Gutnick, M.J. (1977) Extracellular free calcium and potassium during paroxsmal activity in the cerebral cortex of the cat. Experimental Brain Research 27, 237–243.
Hoebe, K. and Beutler, B. (2008) Forward genetic analysis of TLR-signaling pathways: an evaluation. Advanced Drug Delivery Reviews 60, 824–829.
Horton, R.W., Prestwich, S.A. and Meldrum, B.S. (1982) gamma-Aminobutyric acid and benzodiazepine binding sites in audiogenic seizure-susceptible mice. Journal of Neurochemistry 39, 864–870.
Jacobs, K.M., Kharazia, V.N. and Prince, D.A. (1999) Mechanisms underlying epileptogenesis in cortical malformations. Epilepsy Research 36, 165–188.
Jin, X., Huguenard, J.R. and Prince, D.A. (2005) Impaired Cl- extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. Journal of Neurophysiology 93, 2117–2126.
Joels, M. and Baram, T.Z. (2009) The neuro-symphony of stress. Nature Reviews Neuroscience 10, 459–466.
Johnston, D. and Brown, T.H. (1984) The synaptic nature of the paroxysmal depolarizing shift in hippocampal neurons. Annals of Neurology 16 (Suppl), S65–71.
Kabova, R., Liptakova, S., Slamberova, R., Pometlova, M. and Velisek, L. (1999) Age-specific N-methyl-Daspartate-induced seizures: perspectives for the West syndrome model. Epilepsia 40, 1357–1369.
Kelley, K.A., Ho, L., Winger, D., Freire-Moar, J., Borelli, C.B., Aisen, P.S. and Pasinetti, G.M. (1999) Potentiation of excitotoxicity in transgenic mice overexpressing neuronal cyclooxygenase-2. American Journal of Pathology 155, 995–1004.
Khuu, C.H., Barrozo, R.M., Hai, T. and Weinstein, S.L. (2007) Activating transcription factor 3 (ATF3) represses the expression of CCL4 in murine macrophages. Molecular Immunology 44, 1598–1605.
King, R.D., Wiest, M.C. and Montague, P.R. (2001) Extracellular calcium depletion as a mechanism of short-term synaptic depression. Journal of Neurophysiology 85, 1952–1959.
Kirchner, A., Veliskova, J. and Velisek, L. (2006) Differential effects of low glucose concentrations on seizures and epileptiform activity in vivo and in vitro. European Journal of Neuroscience 23, 1512–1522.
Kostopoulos, G., Avoli, M. and Gloor, P. (1983) Participation of cortical recurrent inhibition in the genesis of spike and wave discharges in feline generalized penicillin epilepsy. Brain Research 267, 101–112.
Kovacs, Z., Kekesi, K.A., Szilagyi, N., Abraham, I., Szekacs, D., Kiraly, N., Papp, E., Csaszar, I., Szego, E., Barabas, K., Peterfy, H., Erdei, A., Bartfai, T. and Juhasz, G. (2006) Facilitation of spike-wave discharge activity by lipopolysaccharides in Wistar Albino Glaxo/Rijswijk rats. Neuroscience 140, 731–742.
Kulkarni, S.K. and Dhir, A. (2009) Cyclooxygenase in epilepsy: from perception to application. Drugs Today (Barc) 45, 135–154.
Lee, K.I., Lim, C.Y., Kang, B.T. and Park, H.M. (2011) Clinical and MRI findings of lissencephaly in a mixed breed dog. Journal of Veterinary Medical Science 73, 1385–1388.
Liesz, A., Suri-Payer, E., Veltkamp, C., Doerr, H., Sommer, C., Rivest, S., Giese, T. and Veltkamp, R. (2009) Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nature Medicine 15, 192–199.
Longhi, L., Perego, C., Ortolano, F., Zanier, E.R., Bianchi, P., Stocchetti, N., Mcintosh, T.K. and De Simoni, M.G. (2009) C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Critical Care Medicine 37, 659–665.
Loscher, W. and Schwark, W.S. (1985) Evidence for impaired GABAergic activity in the substantia nigra of amygdaloid kindled rats. Brain Research 339, 146–150.
Loscher, W. and Schwartz-Porsche, D. (1986) Low levels of gamma-aminobutyric acid in cerebrospinal fluid of dogs with epilepsy. Journal of Neurochemistry 46, 1322–1325.
MacVicar, B.A. and Dudek, F.E. (1980) Local synaptic circuits in rat hippocampus: interactions between pyramidal cells. Brain Research 184, 220–223.
Mantovani, A., Locati, M., Vecchi, A., Sozzani, S. and Allavena, P. (2001) Decoy receptors: a strategy to regulate inflammatory cytokines and chemokines. Trends in Immunology 22, 328–336.
Matsumoto, H. and Marsan, C.A. (1964) Cortical Cellular Phenomena in Experimental Epilepsy: Ictal Manifestations. Experimental Neurology 9, 305–326.
Mayberg, H.S., Sadzot, B., Meltzer, C.C., Fisher, R.S., Lesser, R.P., Dannals, R.F., Lever, J.R., Wilson, A.A., Ravert, H.T., Wagner, H.N., Jr et al. (1991) Quantification of mu and non-mu opiate receptors in temporal lobe epilepsy using positron emission tomography. Annals of Neurology 30, 3–11.
Mazarati, A.M. and Wasterlain, C.G. (1999) N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neuroscience Letters 265, 187–190.
McNamara, J.O. (1999) Emerging insights into the genesis of epilepsy. Nature 399, A15–22.
Meisler, M.H., Kearney, J., Ottman, R. and Escayg, A. (2001) Identification of epilepsy genes in human and mouse. Annual Review of Genetics 35, 567–588.
Meldrum, B.S. (1994) The role of glutamate in epilepsy and other CNS disorders. Neurology 44, S14–23.
Meldrum, B.S. and Chapman, A.G. (1999) Excitatory amino acid receptors and antiepileptic drug development. Advances in Neurology 79, 965–978.
Meldrum, B.S., Akbar, M.T. and Chapman, A.G. (1999) Glutamate receptors and transporters in genetic and acquired models of epilepsy. Epilepsy Research 36, 189–204.
Mikati, M.A., Saab, R., Fayad, M.N. and Choueiri, R.N. (2002) Efficacy of intravenous immunoglobulin in Landau-Kleffner syndrome. Pediatric Neurology 26, 298–300.
Mikati, M.A., Kurdi, R., El-Khoury, Z., Rahi, A. and Raad, W. (2010) Intravenous immunoglobulin therapy in intractable childhood epilepsy: open-label study and review of the literature. Epilepsy and Behavior 17, 90–94.
Miles, R. and Wong, R.K. (1983) Single neurones can initiate synchronized population discharge in the hippocampus. Nature 306, 371–373.
Moldrich, R.X., Chapman, A.G., De Sarro, G. and Meldrum, B.S. (2003) Glutamate metabotropic receptors as targets for drug therapy in epilepsy. European Journal of Pharmacology 476, 3–16.
Moody, W.J., Futamachi, K.J. and Prince, D.A. (1974) Extracellular potassium activity during epileptogenesis. Experimental Neurology 42, 248–263.
Mori, A., Hiramatsu, M., Namba, S., Nishimoto, A., Ohmoto, T., Mayanagi, Y. and Asakura, T. (1987) Decreased dopamine level in the epileptic focus. Research Communications in Chemical Pathology and Pharmacology 56, 157–164.
Morita, T., Takahashi, M., Takeuchi, T., Hikasa, Y., Ikeda, S., Sawada, M., Sato, K., Shibahara, T. and Shimada, A. (2005) Changes in extracellular neurotransmitters in the cerebrum of familial idiopathic epileptic shetland sheepdogs using an intracerebral microdialysis technique and immunohistochemical study for glutamate metabolism. Journal of Veterinary Medical Science 67, 1119–1126.
Nadler, J.V. (2003) The recurrent mossy fiber pathway of the epileptic brain. Neurochemical Research 28, 1649–1658.
Nguyen, M.D., Julien, J.P. and Rivest, S. (2002) Innate immunity: the missing link in neuroprotection and neurodegeneration? Nature Reviews Neuroscience 3, 216–227.
Olsen, R.W., Wamsley, J.K., Mccabe, R.T., Lee, R.J. and Lomax, P. (1985) Benzodiazepine/gamma-aminobutyric acid receptor deficit in the midbrain of the seizure-susceptible gerbil. Proceedings of the National Academy of Sciences USA 82, 6701–6705.
Patrylo, P.R., Van Den Pol, A.N., Spencer, D.D. and Williamson, A. (1999) NPY inhibits glutamatergic excitation in the epileptic human dentate gyrus. Journal of Neurophysiology 82, 478–483.
Perkins, N.D. (2007) Integrating cell-signalling pathways with NF-kappaB and IKK function. Nature Reviews – Molecular Cell Biology 8, 49–62.
Pilz, D.T., Matsumoto, N., Minnerath, S., Mills, P., Gleeson, J.G., Allen, K.M., Walsh, C.A., Barkovich, A.J., Dobyns, W.B., Ledbetter, D.H. and Ross, M.E. (1998) LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Human Molecular Genetics 7, 2029–2037.
Pinto, D.J., Patrick, S.L., Huang, W.C. and Connors, B.W. (2005) Initiation, propagation, and termination of epileptiform activity in rodent neocortex in vitro involve distinct mechanisms. Journal of Neuroscience 25, 8131–8140.
Pitkanen, A. and Lukasiuk, K. (2009) Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy and Behavior 14 (Suppl 1), 16–25.
Pitkanen, A. and Sutula, T.P. (2002) Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurology 1, 173–181.
Podell, M. and Hadjiconstantinou, M. (1997) Cerebrospinal fluid gamma-aminobutyric acid and glutamate values in dogs with epilepsy. American Journal of Veterinary Research 58, 451–456.
Podell, M. and Hadjiconstantinou, M. (1999) Low concentrations of cerebrospinal fluid GABA correlate to a reduced response to phenobarbital therapy in primary canine epilepsy. Journal of Veterinary Internal Medicine 13, 89–94.
Przewlocka, B., Lason, W., Machelska, H., Van Luijtelaar, G., Coenen, A. and Przewlocki, R. (1995) Kappa opioid receptor agonists suppress absence seizures in WAG/Rij rats. Neuroscience Letters 186, 131–134.
Rakic, P. (1988) Specification of cerebral cortical areas. Science 241, 170–176.
Ransohoff, R.M., Kivisakk, P. and Kidd, G. (2003) Three or more routes for leukocyte migration into the central nervous system. Nature Reviews - Immunology 3, 569–581.
Ravizza, T. and Vezzani, A. (2006) Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience 137, 301–308.
Ravizza, T., Gagliardi, B., Noe, F., Boer, K., Aronica, E. and Vezzani, A. (2008) Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiology of Disease 29, 142–160.
Rice, A.C. and DeLorenzo, R.J. (1998) NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Research 782, 240–247.
Richerson, G.B. (2004) Looking for GABA in all the wrong places: the relevance of extrasynaptic GABA(A) receptors to epilepsy. Epilepsy Currents 4, 239–242.
Rizzi, M., Perego, C., Aliprandi, M., Richichi, C., Ravizza, T., Colella, D., Veliskova, J., Moshe, S.L., De Simoni, M.G. and Vezzani, A. (2003) Glia activation and cytokine increase in rat hippocampus by kainic acid-induced status epilepticus during postnatal development. Neurobiology of Disease 14, 494–503.
Rogawski, M.A. and Donevan, S.D. (1999) AMPA receptors in epilepsy and as targets for antiepileptic drugs. Advances in Neurology 79, 947–963.
Rothwell, N.J. (1989) CRF is involved in the pyrogenic and thermogenic effects of interleukin 1 beta in the rat. American Journal of Physiology 256, E111–115.
Rutecki, P.A., Lebeda, F.J. and Johnston, D. (1985) Epileptiform activity induced by changes in extracellular potassium in hippocampus. Journal of Neurophysiology 54, 1363–1374.
Rutecki, P.A., Grossman, R.G., Armstrong, D. and Irish-Loewen, S. (1989) Electrophysiological connections between the hippocampus and entorhinal cortex in patients with complex partial seizures. Journal of Neurosurgery 70, 667–675.
Saito, M., Sharp, N.J., Kortz, G.D., De Lahunta, A., Leventer, R.J., Tokuriki, M. and Thrall, D.E. (2002) Magnetic resonance imaging features of lissencephaly in 2 Lhasa Apsos. Veterinary Radiology and Ultrasound 43, 331–337.
Sakurai, M., Morita, T., Takeuchi, T. and Shimada, A. (2013) Relationship of angiogenesis and microglial activation to seizure-induced neuronal death in the cerebral cortex of Shetland Sheepdogs with familial epilepsy. American Journal of Veterinary Research 74, 763–770.
Sapir, T., Cahana, A., Seger, R., Nekhai, S. and Reiner, O. (1999) LIS1 is a microtubule-associated phospho-protein. European Journal of Biochemistry 265, 181–188.
Sapolsky, R., Rivier, C., Yamamoto, G., Plotsky, P. and Vale, W. (1987) Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science 238, 522–524.
Savic, I., Svanborg, E. and Thorell, J.O. (1996) Cortical benzodiazepine receptor changes are related to frequency of partial seizures: a positron emission tomography study. Epilepsia 37, 236–244.
Sayin, U. and Rutecki, P.A. (2003) Group I metabotropic glutamate receptor activation produces prolonged epileptiform neuronal synchronization and alters evoked population responses in the hippocampus. Epilepsy Research 53, 186–195.
Scharfman, H.E. (2007) The neurobiology of epilepsy. Current Neurology and Neuroscience Reports 7, 348–354.
Schwartz, M. and Shechter, R. (2010) Systemic inflammatory cells fight off neurodegenerative disease. Nature Reviews - Neurology 6, 405–410.
Schwindt, P.C., Spain, W.J. and Crill, W.E. (1989) Long-lasting reduction of excitability by a sodium-dependent potassium current in cat neocortical neurons. Journal of Neurophysiology 61, 233–244.
Semple, B.D., Kossmann, T. and Morganti-Kossmann, M.C. (2010) Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. Journal of Cerebral Blood Flow and Metabolism 30, 459–473.
Sloviter, R.S., Zappone, C.A., Harvey, B.D. and Frotscher, M. (2006) Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyper-inhibition in chronically epileptic rats. Journal of Comparative Neurology 494, 944–960.
Snead, O.C., 3rd (1995) Basic mechanisms of generalized absence seizures. Annals of Neurology 37, 146–157.
Somjen, G.G. (2002) Ion regulation in the brain: implications for pathophysiology. Neuroscientist 8, 254–267.
Staley, K.J., Longacher, M., Bains, J.S. and Yee, A. (1998) Presynaptic modulation of CA3 network activity. Nature Neuroscience 1, 201–209.
Stell, B.M., Brickley, S.G., Tang, C.Y., Farrant, M. and Mody, I. (2003) Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proceedings of the National Academy of Sciences USA 100, 14439–14444.
Sutula, T., He, X.X., Cavazos, J. and Scott, G. (1988) Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science 239, 1147–1150.
Sutula, T., Cascino, G., Cavazos, J., Parada, I. and Ramirez, L. (1989) Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Annals of Neurology 26, 321–330.
Swadlow, H.A. (2003) Fast-spike interneurons and feedforward inhibition in awake sensory neocortex. Cerebral Cortex 13, 25–32.
Szekanecz, Z. and Koch, A.E. (2001) Chemokines and angiogenesis. Current Opinion in Rheumatology 13, 202–208.
Thimm, J., Mechler, A., Lin, H., Rhee, S. and Lal, R. (2005) Calcium-dependent open/closed conformations and interfacial energy maps of reconstituted hemichannels. Journal of Biological Chemistry 280, 10646–10654.
Thiry, A., Dogne, J.M., Supuran, C.T. and Masereel, B. (2007) Carbonic anhydrase inhibitors as anticonvulsant agents. Current Topics in Medicinal Chemistry 7, 855–864.
Tian, G.F., Azmi, H., Takano, T., Xu, Q., Peng, W., Lin, J., Oberheim, N., Lou, N., Wang, X., Zielke, H.R., Kang, J. and Nedergaard, M. (2005) An astrocytic basis of epilepsy. Nature Medicine 11, 973–981.
Timofeev, I., Grenier, F. and Steriade, M. (2004) Contribution of intrinsic neuronal factors in the generation of cortically driven electrographic seizures. Journal of Neurophysiology 92, 1133–1143.
Tracey, K.J. (2002) The inflammatory reflex. Nature 420, 853–859.
Traub, R.D., Dudek, F.E., Taylor, C.P. and Knowles, W.D. (1985) Simulation of hippocampal afterdischarges synchronized by electrical interactions. Neuroscience 14, 1033–1038.
Traub, R.D., Borck, C., Colling, S.B. and Jefferys, J.G. (1996) On the structure of ictal events in vitro. Epilepsia 37, 879–891.
Traub, R.D., Michelson-Law, H., Bibbig, A.E., Buhl, E.H. and Whittington, M.A. (2004) Gap junctions, fast oscillations and the initiation of seizures. Advances in Experimental Medicine and Biology 548, 110–122.
Trevelyan, A.J., Sussillo, D. and Yuste, R. (2007) Feedforward inhibition contributes to the control of epileptiform propagation speed. Journal of Neuroscience 27, 3383–3387.
Trist, D.G. (2000) Excitatory amino acid agonists and antagonists: pharmacology and therapeutic applications. Pharmaceutica Acta Helvetiae 74, 221–229.
Vaillend, C., Mason, S.E., Cuttle, M.F. and Alger, B.E. (2002) Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region. Journal of Neurophysiology 88, 2963–2978.
Velisek, L. (1998) Extracellular acidosis and high levels of carbon dioxide suppress synaptic transmission and prevent the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Hippocampus 8, 24–32.
Velisek, L., Moshe, S.L. and Cammer, W. (1993) Developmental changes in seizure susceptibility in carbonic anhydrase II-deficient mice and normal littermates. Brain Research - Developmental Brain Research 72, 321–324.
Velisek, L., Dreier, J.P., Stanton, P.K., Heinemann, U. and Moshe, S.L. (1994) Lowering of extracellular pH suppresses low-Mg(2+)-induces seizures in combined entorhinal cortex-hippocampal slices. Experimental Brain Research 101, 44–52.
Verhelst, H., Boon, P., Buyse, G., Ceulemans, B., D’Hooghe, M., Meirleir, L.D., Hasaerts, D., Jansen, A., Lagae, L., Meurs, A., Coster, R.V. and Vonck, K. (2005) Steroids in intractable childhood epilepsy: clinical experience and review of the literature. Seizure 14, 412–421.
Vezzani, A. and Baram, T.Z. (2007) New roles for interleukin-1 Beta in the mechanisms of epilepsy. Epilepsy Currents 7, 45–50.
Vezzani, A. and Granata, T. (2005) Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46, 1724–1743.
Vezzani, A., Conti, M., De Luigi, A., Ravizza, T., Moneta, D., Marchesi, F. and De Simoni, M.G. (1999) Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. Journal of Neuroscience 19, 5054–5065.
Vezzani, A., Moneta, D., Conti, M., Richichi, C., Ravizza, T., De Luigi, A., De Simoni, M.G., Sperk, G., Andell-Jonsson, S., Lundkvist, J., Iverfeldt, K. and Bartfai, T. (2000a) Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proceedings of the National Academy of Sciences USA 97, 11534–11539.
Vezzani, A., Rizzi, M., Conti, M. and Samanin, R. (2000b) Modulatory role of neuropeptides in seizures induced in rats by stimulation of glutamate receptors. Journal of Nutrition 130, 1046S–1048S.
Vezzani, A., French, J., Bartfai, T. and Baram, T.Z. (2011) The role of inflammation in epilepsy. Nature Reviews Neurology 7, 31–40.
Vezzani, A., Aronica, E., Mazarati, A. and Pittman, Q.J. (2013) Epilepsy and brain inflammation. Experimental Neurology 244, 11–21.
Vincent, A. and Bien, C.G. (2008) Anti-NMDA-receptor encephalitis: a cause of psychiatric, seizure, and movement disorders in young adults. Lancet Neurology 7, 1074–1075.
Vincent, A., Irani, S.R. and Lang, B. (2010) The growing recognition of immunotherapy-responsive seizure disorders with autoantibodies to specific neuronal proteins. Current Opinion in Neurology 23, 144–150.
Virta, M., Hurme, M. and Helminen, M. (2002) Increased plasma levels of pro- and anti-inflammatory cytokines in patients with febrile seizures. Epilepsia 43, 920–923.
Wheless, J.W., Clarke, D.F., Arzimanoglou, A. and Carpenter, D. (2007) Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disorders 9, 353–412.
Wilson, E.H., Weninger, W. and Hunter, C.A. (2010) Trafficking of immune cells in the central nervous system. Journal of Clinical Investigation 120, 1368–1379.
Wirrell, E., Farrell, K. and Whiting, S. (2005) The epileptic encephalopathies of infancy and childhood. Canadian Journal of Neurological Science 32, 409–418.
Wong, R.K. and Watkins, D.J. (1982) Cellular factors influencing GABA response in hippocampal pyramidal cells. Journal of Neurophysiology 48, 938–951.
Wu, Y., Wang, X., Mo, X., Xi, Z., Xiao, F., Li, J., Zhu, X., Luan, G., Wang, Y., Li, Y. and Zhang, J. (2008) Expression of monocyte chemoattractant protein-1 in brain tissue of patients with intractable epilepsy. Clinical Neuropathology 27, 55–63.
Xiong, Z.Q., Qian, W., Suzuki, K. and Mcnamara, J.O. (2003) Formation of complement membrane attack complex in mammalian cerebral cortex evokes seizures and neurodegeneration. Journal of Neuroscience 23, 955–960.
Xu, J.H., Long, L., Tang, Y.C., Zhang, J.T., Hut, H.T. and Tang, F.R. (2009) CCR3, CCR2A and macrophage inflammatory protein (MIP)-1a, monocyte chemotactic protein-1 (MCP-1) in the mouse hippocampus during and after pilocarpine-induced status epilepticus (PISE). Neuropathology and Applied Neurobiology 35, 496–514.
Yamada, K., Ji, J.J., Yuan, H., Miki, T., Sato, S., Horimoto, N., Shimizu, T., Seino, S. and Inagaki, N. (2001) Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science 292, 1543–1546.
Yoshikawa, K., Kita, Y., Kishimoto, K. and Shimizu, T. (2006) Profiling of eicosanoid production in the rat hippocampus during kainic acid-induced seizure: dual phase regulation and differential involvement of COX-1 and COX-2. Journal of Biological Chemistry 281, 14663–14669.
You, S.J., Jung, D.E., Kim, H.D., Lee, H.S. and Kang, H.C. (2008) Efficacy and prognosis of a short course of prednisolone therapy for pediatric epilepsy. European Journal of Paediatric Neurology 12, 314–320.
2 Pathophysiology of Pharmacoresistant
Epilepsy
Holger A. Volk
Professor, Diplomate of the European College of Veterinary Neurology, Department of Clinical Science and Services, The Royal Veterinary College, UK
Introduction
Epilepsy is the most common chronic neurological condition in people with an estimated incidence of 0.05–0.1% and prevalence of 0.4–1% (Sander and Shorvon, 1996; Cowan, 2002). Despite treatment with two adequate antiepileptic drugs (AEDs), 23% of human patients continue to have seizures (Picot et al., 2008). Epilepsy has also been suggested in dogs to be the most common chronic neurological disorder (Chandler, 2006; Fluehmann et al., 2006) with an estimated prevalence of 1–2% in a referral hospital population (SchwartzPorsche, 1986) and 0.6% in first opinion practice (KearsleyFleet et al., 2013). Around 75–85% of dogs with idiopathic epilepsy will continue to have seizures (Heynold et al., 1997; Berendt et al., 2002, 2007; Arrol et al., 2012) and around 20–30% will remain poorly controlled (<50% reduction of seizure frequency) despite adequate treatment with phenobarbitone (PB) and/or potassium bromide (KBr) (SchwartzPorsche et al., 1985; Podell and Fenner, 1993; Trepanier et al., 1998). Thus, with an estimated current UK companion dog population of
9.4 million, around 55,000 dogs have idiopathic epilepsy of which 12500 will be classified as poorly controlled. Dogs with epilepsy, especially poorly controlled epilepsy with a high seizure frequency, have an increased risk of premature death, behaviour changes and a reduced quality of life (Chang et al., 2006; Berendt et al., 2007; Shihab et al., 2011; Wessmann et al., 2012). Seizures do not only affect the quality of life for the affected dogs, but also for the pet owner (Chang et al., 2006; Wessmann et al., 2012). Improving our understanding of why some patients respond to treatment while others do not is therefore of key importance.
Epilepsy is caused by a heterogeneous group of chronic conditions with seizures being its clinical manifestation. Despite the brain’s complex structure, there are only limited ways in which it can demonstrate its function and dysfunction. A wide variety of disturbances of brain structure and function can result in seizures and epilepsy. However, similar pathophysiological pathways can lead to other neurodevelopmental and neurobehavioural disorders (Johnson and Shorvon, 2011; Shihab et al., 2011). Many of these disturbances share commonalities at various levels, which can help to understand, diagnose, monitor and treat these disorders.
Investigations into why treatment could fail can therefore:
• Shed light on the mechanisms causing a refractory state to drug therapy and how to overcome them; and
Pathophysiology of Pharmacoresistant Epilepsy
• Deepen our understanding of the pathophysiology of epilepsy and its natural clinical course.
Definition
The definition of pharmacoresistant epilepsy has been a matter of debate in human medicine. Formerly, epilepsy was considered responsive to medical treatment if seizures were reduced by ³50% to one or two adequate AED which achieved therapeutic serum concentration and steady state (Regesta and Tanganelli, 1999). Most current epilepsy drug trials in veterinary medicine have used this definition to determine AED efficacy (Dewey et al., 2004, 2009; Platt et al., 2006; von Klopmann et al., 2007; Volk et al., 2008; Muñana et al., 2010, 2012b). However, this does not take into account that even a reduction of seizure frequency by more than 50% may still entail a high seizure frequency, which negatively impacts on the animal’s and the owner’s quality of life (Chang et al., 2006; Wessmann et al., 2012). A good quality of life is however best improved by seizure freedom, one of the main outcome measures nowadays in epilepsy trials in human medicine (Elsharkawy et al., 2009).
A task force of the International League against Epilepsy (ILAE) has recently agreed on a global consensus on granted outcome measures for therapeutic interventions (Level 1) and definition of pharmacoresistant epilepsy (Level 2) taking the occurrence of druginduced side effects and seizure control into account (Kwan et al., 2010):
• Level 1 categorizes the outcome of each medical treatment as either free of seizures or treatment failure, if not seizure free. The ILAE recommends a followup time of three times the longest interictal period for therapeutic trials; e.g. if the period in between seizures is 1 month then the followup time should be at least 3 months. However, as this is not always practical, seizure freedom is usually defined as an absence of seizures for a 12month period. Level 1 does not only consider efficacy, but also the occurrence of druginduced side effects, which will contribute significantly to the overall impact on the patient’s quality of life.
• Level 2 defines pharmacoresistant epilepsy as a failure to achieve freedom from seizures despite adequate trials of two (or more) welltolerated, correctly chosen and used AED regimens (whether administered as monotherapies or in combination) (Kwan et al., 2010). The number of AED trialled has been restricted to ‘two’ based on the observation that successful seizure control is very unlikely once the patient has not responded to two correctly chosen AED (Kwan and Brodie, 2000; Arts et al., 2004).
It needs to be considered that drug nonresponsiveness is not a fixed state but rather a dynamic process, which can be altered by fluctuation of the underlying pathophysiology (Kwan et al., 2010). The seizure frequency can vary over time (Berg et al., 2009; Muñana et al., 2010) and the state of drug nonresponsiveness is dependent on the timepoint of assessment (Kwan et al., 2010).
Taking into consideration the revised ILAE classification of pharmacoresistant epilepsy, a high proportion of dogs would be classified as pharmacoresistant. Spontaneous and druginduced epilepsy remission rate in people is around 63% (Kwan and Brodie, 2000), which is markedly higher than that reported in veterinary medicine, which usually ranges between 15 and 24% (Heynold et al., 1997; Berendt et al., 2007; Arrol et al., 2012). However, in a recent study comparing the efficacy and tolerability of phenobarbitone with potassium bromide as a first line treatment, seizures ceased in 85% and 52% of the treated dogs, respectively (Boothe et al., 2012). The study period was only 6 months long and thus it is likely that the percentage of dogs being seizure free would have been less with a longer followup period. Lagotto Romagnolo dogs have a documented ‘childhood’ or juvenile epilepsy, which starts when they are around 6 weeks of age (Jokinen et al., 2007). Of the affected dogs, 96% become seizure free at the age of 10 weeks generally without receiving AED treatment. Future, longterm epidemiological studies are necessary to determine prevalence of drug response more accurately in the general epileptic dog population, in addition to the effect of patient age and duration of seizure history on this response.
Risk Factors for Pharmacoresistant Epilepsy
Genetic risk factors
An increased prevalence of epilepsy has been described for many dog breeds (Jaggy et al., 1998; Kathmann et al., 1999; Berendt et al., 2002, 200; Casal et al., 2006; Gullov et al., 2011), which raises the possibility of genetic risk factors also being responsible for drugresponsiveness. The Lagotto Romagnolo, as aforementioned, has a benign childhood or juvenile epilepsy, which goes into remission in many affected dogs with age, as the function of the Lgi2 gene in cerebral synaptic remodelling becomes less important (Jokinen et al., 2007; Seppala et al., 2011). The Lgi2 gene is involved in the immediate postnatal phase of neuronal pruning, but then the Lgi1 gene has a more important role in the regulation of the later part of pruning.
Another gene mutation that has been associated with improved seizure control is located on the ABCB1 gene. The ABCB1 (multidrug resistance (MDR) 1) gene encodes a transmembrane protein, the permeabilityglycoprotein (Pgp). Pgp, an ATPdependent multidrug transporter, expressed at the blood–brain barrier (BBB), protects the brain from potential central nervous system (CNS) toxins (Schinkel et al., 1994, 1996; Schrickx and FinkGremmels, 2008), including AEDs such as PB (Potschka et al., 2002; Basic et al., 2008). Dysfunction of this neuroprotective BBB efflux pump can lead to BBB dysfunction and consequently CNS intoxication (Schinkel et al., 1994, 1996). Collies and other herding breeds can be affected by such a dysfunction of the Pgp transporter caused by a four basepair deletion (c.296_ 299del) in exon 4 of the ABCB1 gene (Mealey et al., 2001). A homozygous deletion in this region cannot only lead to CNS intoxication by drugs such as ivermectin, but also has been suggested to improve medical seizure control in affected collies (Muñana et al., 2012a).
Border collies are, however, known to have an aggressive seizure phenotype characterized by high seizure frequency, cluster seizures and poor drug responsiveness. In a recent study, 71% of Border collies were classified as pharmacoresistant (Hülsmeyer et al., 2010). PBresistant epileptic Border collies had a single nucleotide polymorphism (SNP) at intron 1 near the 5end of the ABCB1 gene, which could influence the promoter elements of this gene (Alves et al., 2011). A polymorphism at the promoter region could influence transcription activity and therefore might increase the level of Pgp expression at the BBB. The same group looked at another breed, the Australian shepherd dog, which is closely related to the Border collie and also has a severe epilepsy phenotype (Weissl et al., 2012). In this dog breed, they were not able to identify a polymorphism related to drugrefractoriness.
Another recent study looked at gene differences between PB responders and nonresponders (Kennerly et al., 2009). Five genes were suggested to have an association with PB response, although they did not reach statistical significance after adjustment for multiple comparisons (KCNQ3, SCN2A, GABRA2, EPOX HYD and ABCB4). Three of these genes encode ion channels (KCNQ3, voltage gated potassium ion channel important for postexcitatory membrane repolarization; SCN2A, sodium ion channel; GABRA2, GABA receptor), all of them are AED targets (Armijo et al., 2005). The other two genes are involved in PB metabolism (EPOX HYD) or transportation (ABCB4) (Kennerly et al., 2009).
Clinical risk factors
Apart from identifying genetic backgrounds, which can be associated with poor response to AEDs, much attention has been spent in the last decade to identify clinical risk factors that predict pharmacoresistant epilepsy. In people, it was commonly believed that seizure freedom was more likely to be achieved when a patient received AEDs immediately
Pathophysiology of Pharmacoresistant Epilepsy
after the occurrence of the first seizure. Interestingly, epidemiological studies in developing countries, where AEDs are not freely available, revealed remission rates similar to those in industrial countries (Placencia et al., 1993). Most AEDs despite having good seizure suppressing activities have little influence on the natural course of the disease (epilepsy) itself. New AEDs that were thought to also have an antiepileptogenic effect have failed in chronic epilepsy models and have not prevented the development of epilepsy (Brandt et al., 2007).
Heynold and colleagues have shown in 1997 that Labrador retrievers, which achieved freedom from seizures, received medication a longer period of time after their first seizure than those dogs that continued to seizure (Heynold et al., 1997). This implies that timing in relation to onset of seizure activity does not influence longterm seizure control in the canine patient. However, there is enough evidence in dogs, rodent models and humans that disease severity, such as high seizure frequency, cluster seizures and status epilepticus can influence drug responsiveness (Heynold et al., 1997; Kwan and Brodie, 2000; Hülsmeyer et al., 2010; Löscher and Brandt, 2010; Weissl et al., 2012). Intact male and female dogs have a higher likelihood of having cluster seizures (Monteiro et al., 2012). Seizure severity will ultimately guide clinical reasoning and the more severely affected patient will receive treatment earlier than dogs with a less severe epilepsy phenotype. It was formerly believed that very young dogs starting to have recurrent seizures have a worse outcome. As an example, Labrador retrievers had a better outcome when they developed epilepsy later in life (Heynold et al., 1997). However, a recent study from the UK could not find that the onset of seizures before the age of 1 year had any influence on survival outcome (Arrol et al., 2012). Interestingly, the same group could not find that the presence of focal seizures influenced the overall outcome, which is different to what is seen in human medicine (Kwan et al., 2011).
Pseudoresistance
Once a clinician encounters pharmacoresistant epilepsy, the first question to be asked should be about the possibility of an identifiable, underlying disease process. If seizures persist because the underlying disease has not been identified and treated incorrectly, it is termed pseudoresistance. Conditions mimicking epilepsy (see Chapter 9) need to be also considered; these include cardiac associated syncope, transient vestibular disorders, movement disorders and episodic pain. Sometimes AED treatment can aggravate such mimics (Penning et al., 2009) and so each patient presenting with assumed pharmacoresistant epilepsy needs to be thoroughly investigated with an open mind. One may also encounter failure of AED therapy if owner compliance is poor or pharmacokinetic and dynamic properties were not considered adequately.
Mechanisms of Pharmacoresistant Epilepsy
There are many possible causes of pharmacoresistant epilepsy. The mechanisms of drug resistance are likely to be variable and multifactorial (Regesta and Tanganelli, 1999; Kwan et al., 2011). Genetic factors, such as the aforementioned polymorphisms, may be relevant and help to explain why two dogs of the same breed and with the same epilepsy characteristics differ in their response to AED therapy. Diseaserelated factors are also of importance, and these include the aetiology and pathophysiology underlying the seizure disorder, the clinical course of the disease, changes in drug targets or binding sites, drug uptake into the brain and changes of the epilepsy circuit. Finally, drugrelated factors such as ineffective pharmacodynamics or kinetic properties and/or development of tolerance can all play a role in the formation of pharmacoresistance.
Most pharmacoresistant patients will often not respond to multiple AEDs despite the individual differences in their mechanisms of action (Regesta and Tanganelli, 1999; Löscher and Potschka, 2002). Patients that do not respond to the first standard AED will only have a 3% chance to respond to the subsequent chosen AEDs, which needs especially to be considered during AED trials (Kwan and Brodie, 2000). This argues against epilepsyinduced changes in specific drug targets as a major cause of drugresistant epilepsy, and makes it more likely that nonspecific mechanisms are responsible.
There are three proposed major theories for AED resistance:
Drug-target hypothesis
Based on the drug target hypothesis, reduced sensitivity of drug targets such as receptors or ion channels to AEDs is a key cellular mechanism that may cause drug resistance. This hypothesis is based on findings that show that in the hippocampus of human patients with pharmacoresistant temporal lobe epilepsy, the usedependent inhibition of sodium channels by carbamazepine is lost. This finding did not extend to lamotrigine, which has a pharmacologic action similar to that of carbamazepine (Remy et al., 2003). Polymorphisms in the sodium channel encoding gene SCN2A were found in humans to be responsible for AEDs acting on the sodium channel, but also for other nonsodium channel targeted AEDs (Kwan et al., 2008). As aforementioned, Kennerly et al. (2009) showed that PB nonresponders also had changes in the SCN2A gene when compared to PB responders. In addition, two other genes encoding ion channels (KCNQ3 and GABRA2) were affected (Armijo et al., 2005). In a rodent model for pharmacoresistant epilepsy it was demonstrated that there was a shift from GABAA diazepamsensitive to GABAA diazepaminsensitive receptors in the hippocampus of PB nonresponders (Volk et al., 2006). In the same model a significant loss of neurons in the CA1, CA3c/CA4 and dentate hilus of nonresponders was found. This could lead to altered network properties, which could be responsible for refractoriness. In human medicine, altered network properties are well recognized in patients with hippocampal sclerosis. Hippocampal sclerosis has been associated with refractoriness to AEDs (Kwan et al., 2011). In dogs, the hippocampus is also involved in seizure propagation, which can result in MRI changes (Kuwabara et al., 2010). Such MRI abnormalities have not been associated with changes in seizure frequency or length in dogs but hippocampal changes visible on MRI are often associated with AED therapy failure in cats (Fatzer et al., 2000; Brini et al., 2004; Schmied et al., 2008).
A wealth of literature on human epilepsy cites evidence of autoantibodies to ion channels (GABAB receptors, Lancaster et al., 2010; NMDAreceptors, Dalmau et al., 2008; calcium channels, McKnight et al., 2005; voltagegated potassium channels, McKnight et al., 2005). Interestingly, it also appears that cats with hippocampal changes develop autoantibodies to voltagegated potassium channels (Pakozdy et al., 2013). These patients rarely respond to standard AED and in human medicine immunomodulatory treatment has been trialled with variable results (Vincent et al., 2010). Other proposed cellular pathomechanisms of pharmacoresistant epilepsy that have been suggested include electrical coupling via gap junctions in neurons and glial cells (Voss et al., 2009) and mitochondrial oxidative stress and dysfunction (Waldbaum and Patel, 2010).
Multidrug transporter hypothesis
A drug can only enter the brain via two routes, either traversing the BBB or via the ventricular system and cerebrospinal fluid (CSF). The BBB restricts the entrance of any drug or other xenobiotic substance in order to protect the CNS from toxicity. The endothelial cells in the BBB are connected by tight junctions and surrounded by a basement membrane with astrocytic foot processes covering 95% of the endothelial lining. The BBB lacks transendothelial pathways such as transcellular channels or fenestrations (Löscher and Potschka, 2002). The functional significance is that the BBB resembles continuous phospholipid membranes, which stops the diffusion of hydrophilic, large or proteinbound drugs.
Pathophysiology of Pharmacoresistant Epilepsy
Lipidsoluble drugs were thought on the other hand to diffuse easily through the BBB. Apart from the passive transport mechanism of lipophilic compounds, the BBB hosts a carriermediated transport system (Pardridge, 1999). In the last decade, multiple multidrug transporters of the ATPbinding cassette superfamily, especially Pgp and multidrug resistanceassociated protein (MRP), have been shown to be expressed physiologically on the luminal side of the endothelial cells of the BBB. Pgp and MRP are also expressed in the choroid plexus epithelial cells limiting brain entrance even further (Rao et al., 1999). These transporters appear to act as an active defence mechanism against the penetration of potential CNS toxic lipophilic compounds, therefore limiting the penetration of lipophilic drugs, such as AEDs (Fromm, 2000; Spector, 2000). AEDs which are transported by multidrug transporters include valproate, gabapentin, topiramate, phenytoin, carbamazepine, phenobarbitone, felbamate and lamotrigine (Löscher and Potschka, 2002).
In contrast to the aforementioned hypotheses of drugrefractoriness, the multidrug transporter hypothesis is based on the assumption that it is not the brain target itself but the reduced AED concentration at the target site that causes pharmacoresistance. Indeed, an increased expression of multidrug transporters, such as Pgp and MRP in brain capillary endothelial cells comprising the BBB has been demonstrated both in epileptogenic brain tissue of human pharmacoresistant patients and in animal models of pharmacoresistant epilepsy (Tishler et al., 1995; Sisodiya et al., 2002; Aronica et al., 2003; Potschka et al., 2004; Volk et al., 2004a, b; Volk and Löscher, 2005; Hoffmann et al., 2006). In pharmacoresistant epileptic tissue, Pgp is also overexpressed in astrocytic foot processes and neurons limiting the AED concentration at the target even further (Aronica et al., 2003; Volk et al., 2004a). Overexpression of the multidrug transporter Pgp was demonstrated in dogs after increased seizure activity, status epilepticus and cluster seizures (Pekcec et al., 2009b). This is an interesting finding as seizure frequency and severity are, as aforementioned, clinical risk factors for pharmacoresistant epilepsy.
More than 50 SNPs and insertion/deletion polymorphisms have been reported for ABCB1, with some of them resulting in a change of Pgp expression and/or function (Löscher and Potschka, 2005). An aforementioned study has also described a polymorphism in the promoter region of ABCB1 in Border collies associated with poor controlled epilepsy (Alves et al., 2011). This polymorphism could also have resulted in a change of function or overexpression of Pgp. However, no functional study was performed. The authors also found three other SNPs in the coding region of ABCB1, which were not associated with drug response or epilepsy. Apart from genetic influences, druginduced mechanisms will interfere with multidrug transporter expression at the BBB. The BBB efflux multidrug transporter expression adapts continuously to ensure protection and detoxification of the CNS from xenobiotic substances. Efflux transporter expression is regulated by pregnane X receptor (PXR), which reacts to xenobiotic (foreign toxic) compound exposure (Masuyama et al., 2005; Miller, 2010; Shukla et al., 2011). This results in a clearing of these compounds from the brain and/or body. In addition to the upregulation of multidrug transporter expression, PXR coregulates drug metabolizing CytP450 enzymes (Potschka, 2012). The binding domain of PXR and Pgp have many similarities and will interact with similar compounds, resulting in a dynamic process of regulating the efflux of xenobiotic compounds. AED have been reported to cause an upregulation of Pgp expression via PXR activation. However, a clear interaction of PXR with standard AEDs remains contentious (Potschka, 2012). It appears that the main driving force for Pgp overexpression is seizure activity (Potschka, 2010). Overexpression is transient and includes brain regions involved in seizure initiation and propagation (Kwan et al., 2002). A high seizure frequency therefore results in an accumulation of efflux transporters at the BBB.
Each epileptic seizure results in a glutamate release, which activates an intracellular signalling cascade in BBB endothelial cells (Bankstahl et al., 2008; Bauer et al., 2008). The glutamate binds to endothelial NmethylDaspartate (NMDA) receptors, which starts arachidonic acid signalling. Cyclooxygenase2 (COX2) processes the arachidonic acid and produces prostaglandin E2 (PGE2). PGE2 binds on the prostaglandin E receptor (EP1) resulting in Pgp expression (Pekcec et al., 2009a).
Should Pgp overexpression be one of the major reasons for drug refractoriness, blocking or reducing the expression of Pgp could reverse the lack of drug response. Several case reports in human medicine and studies in animal models of refractory epilepsy have shown an improved seizure control when Pgp inhibitors were used (Brandt et al., 2006; Potschka, 2012). However, a recent study performed in dogs using verapamil as a Pgp inhibitor did not show a significant reduction of the seizure frequency. Verapamil is not a very specific Pgp inhibitor, which could explain the negative results. Furthermore, verapamil dosage was limited due to its cardiovascular side effects. The other problem with using a Pgp inhibitor is the lack of specificity for the BBB, such that the rest of the excretory body system will be limited in function, therefore the longterm safety of this treatment approach needs to be questioned. A more promising route might be the use of COX2 inhibitors as they have been shown to decrease Pgp in rodent epilepsy studies that also resulted in a reversal of the drug refractoriness (Potschka, 2012).
Conclusion
In conclusion, our understanding of pharmacoresistance has grown significantly over the last several years. Focusing on seizure freedom and quality of life will change the approach in veterinary medicine. In conjunction with a better understanding of mechanisms involved in drug refractoriness a new era of treatment development will have to evolve, hopefully improving animals’ welfare.
References
Alves, L., Hülsmeyer, V., Jaggy, A., Fischer, A., Leeb, T. and Drögemüller, M. (2011) Polymorphisms in the ABCB1 gene in phenobarbital responsive and resistant idiopathic epileptic Border Collies. Journal of Veterinary Internal Medicine 25, 484–489.
Armijo, J.A., Shushtarian, M., Valdizan, E.M., Cuadrado, A., de las Cuevas, I. and Adin, J. (2005) Ion channels and epilepsy. Current Pharmaceutical Design 11, 1975–2003.
Aronica, E., Gorter, J.A., Jansen, G.H., van Veelen, C.W., van Rijen, P.C., Leenstra, S., Ramkema, M., Scheffer, G.L., Scheper, R.J. and Troost, D. (2003) Expression and cellular distribution of multidrug transporter proteins in two major causes of medically intractable epilepsy: focal cortical dysplasia and glioneuronal tumors. Neuroscience 118, 417–429.
Arrol, L., Penderis, J., Garosi, L., Cripps, P., Gutierrez-Quintana, R. and Goncalves, R. (2012) Aetiology and long-term outcome of juvenile epilepsy in 136 dogs. Veterinary Record 170, 335.
Arts, W.F., Brouwer, O.F., Peters, A.C., Stroink, H., Peeters, E.A., Schmitz, P.I., van Donselaar, C.A. and Geerts, A.T. (2004) Course and prognosis of childhood epilepsy: 5-year follow-up of the Dutch study of epilepsy in childhood. Brain 127, 1774–1784.
Bankstahl, J.P., Hoffmann, K., Bethmann, K. and Löscher, W. (2008) Glutamate is critically involved in seizure- induced overexpression of P-glycoprotein in the brain. Neuropharmacology 54, 1006–1016.
Basic, S., Hajnsek, S., Bozina, N., Filipcic, I., Sporis, D., Mislov, D. and Posavec, A. (2008) The influence of C3435T polymorphism of ABCB1 gene on penetration of phenobarbital across the blood-brain barrier in patients with generalized epilepsy. Seizure 17, 524–530.
Bauer, B., Hartz, A.M., Pekcec, A., Toellner, K., Miller, D.S. and Potschka, H. (2008) Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Molecular Pharmacology 73, 1444–1453.
Berendt, M., Gredal, H., Pedersen, L.G., Alban, L. and Alving, J. (2002) A cross-sectional study of epilepsy in Danish Labrador Retrievers: prevalence and selected risk factors. Journal of Veterinary Internal Medicine 16, 262–268.
Berendt, M., Gredal, H., Ersbøll, A.K. and Alving, J. (2007) Premature death, risk factors, and life patterns in dogs with epilepsy. Journal of Veterinary Internal Medicine 21, 754–759.
Pathophysiology of Pharmacoresistant Epilepsy
Berendt, M., Gulløv, C.H., Christensen, S.L.K., Gudmundsdottir, H., Gredal, H., Fredholm, M. and Alban, L. (2008) Prevalence and characteristics of epilepsy in the Belgian shepherd variants Groenendael and Tervueren born in Denmark 1995-2004. Acta Veterinaria Scandinavica 50, 51.
Berg, A.T., Levy, S.R., Testa, F.M. and DSouza, R. (2009) Remission of epilepsy after two drug failures in children: a prospective study. Annals of Neurology 65, 510–509.
Boothe, D.M., Dewey, C. and Carpenter, D.M. (2012) Comparison of phenobarbital with bromide as a first-choice antiepileptic drug for treatment of epilepsy in dogs. Journal of the American Veterinary Medical Association 240, 1073–1083.
Brandt, C., Bethmann, K., Gastens, A.M. and Löscher, W. (2006) The multidrug transporter hypothesis of drug resistance in epilepsy: Proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiology of Disease 24, 202–211.
Brandt, C., Glien, M., Gastens, A.M., Fedrowitz, M., Bethmann, K., Volk, H.A., Potschka, H. and Löscher, W. (2007) Prophylactic treatment with levetiracetam after status epilepticus: lack of effect on epileptogenesis, neuronal damage, and behavioral alterations in rats. Neuropharmacology 53, 207–221.
Brini, E., Gandini, G., Crescio, I., Fatzer, R. and Casalone, C. (2004) Necrosis of hippocampus and piriform lobe: clinical and neuropathological findings in two Italian cats. Journal of Feline Medicine and Surgery 6, 377–381.
Casal, M.L., Munuve, R.M., Janis, M.A., Werner, P. and Henthorn, P.S. (2006) Epilepsy in Irish Wolfhounds. Journal of Veterinary Internal Medicine 20, 131–135.
Chandler, K. (2006) Canine epilepsy: what can we learn from human seizure disorders? The Veterinary Journal 172, 207–217.
Chang, Y., Mellor, D.J. and Anderson, T.J. (2006) Idiopathic epilepsy in dogs: owners’ perspectives on management with phenobarbitone and/or potassium bromide. Journal of Small Animal Practice 47, 574–581.
Cowan, L.D. (2002) The epidemiology of the epilepsies in children. Mental Retardation and Developmental Disabilities Research Reviews 8, 171–181.
Dalmau, J., Gleichman, A.J., Hughes, E.G., Rossi, J.E., Peng, X., Lai, M., Dessain, S.K., Rosenfeld, M.R., Balice-Gordon, R. and Lynch, D.R. (2008) Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurology 7, 1091–1098.
Dewey, C.W., Guiliano, R., Boothe, D.M., Berg, J.M., Kortz, G.D., Joseph, R.J. and Budsberg, S.C. (2004) Zonisamide therapy for refractory idiopathic epilepsy in dogs. Journal of the American Animal Hospital Association 40, 285–291.
Dewey, C.W., Cerda-Gonzalez, S., Levine, J.M., Badgley, B.L., Ducoté, J.M., Silver, G.M., Cooper, J.J., Packer, R.A. and Lavely, J.A. (2009) Pregabalin as an adjunct to phenobarbital, potassium bromide, or a combination of phenobarbital and potassium bromide for treatment of dogs with suspected idiopathic epilepsy. Journal of the American Veterinary Medical Association 235, 1442–1449.
Elsharkawy, A.E., May, T., Thorbecke, R. and Ebner, A. (2009) Predictors of quality of life after resective extratemporal epilepsy surgery in adults in long-term follow-up. Seizure 18, 498–503.
Fatzer, R., Gandini, G., Jaggy, A., Doherr, M. and Vandevelde, M. (2000) Necrosis of hippocampus and piriform lobe in 38 domestic cats with seizures: a retrospective study on clinical and pathologic findings. Journal of Veterinary Internal Medicine 14, 100–104.
Fluehmann, G., Doherr, M.G. and Jaggy, A. (2006) Canine neurological diseases in a referral hospital population between 1989 and 2000 in Switzerland. Journal of Small Animal Practice 47, 582–587.
Fromm, M.F. (2000) P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs. International Journal of Clinical Pharmacology and Therapeutics 38, 69–74.
Gullov, C.H., Toft, N., Baadsager, M.M. and Berendt, M. (2011) Epilepsy in the Petit Basset Griffon Vendeen: prevalence, semiology, and clinical phenotype. Journal of Veterinary Internal Medicine 25, 1372–1378.
Heynold, Y., Faissler, D., Steffen, F. and Jaggy, A. (1997) Clinical, epidemiological and treatment results of idiopathic epilepsy in 54 labrador retrievers: a long-term study. Journal of Small Animal Practice 38, 7–14.
Hoffmann, K., Gastens, A.M., Volk, H.A. and Löscher, W. (2006) Expression of the multidrug transporter MRP2 in the blood-brain barrier after pilocarpine-induced seizures in rats. Epilepsy Research 69, 1–14.
Hülsmeyer, V., Zimmermann, R., Brauer, C., Sauter-Louis, C. and Fischer, A. (2010) Epilepsy in Border Collies: clinical manifestation, outcome, and mode of inheritance. Journal of Veterinary Internal Medicine 24, 171–178.
Jaggy, A., Faissler, D., Gaillard, C., Srenk, P. and Graber, H. (1998) Genetic aspects of idiopathic epilepsy in Labrador retrievers. Journal of Small Animal Practice 39, 275–280.
Johnson, M.R. and Shorvon, S.D. (2011) Heredity in epilepsy: neurodevelopment, comorbidity, and the neurological trait. Epilepsy and Behavior 22, 421–427.
Jokinen, T.S., Metsähonkala, L., Bergamasco, L., Viitmaa, R., Syrjä, P., Lohi, H., Snellman, M., Jeserevics, J. and Cizinauskas, S. (2007) Benign familial juvenile epilepsy in Lagotto Romagnolo dogs. Journal of Veterinary Internal Medicine 21, 464–471.
Kathmann, I., Jaggy, A., Busato, A., Bartschi, M. and Gaillard, C. (1999) Clinical and genetic investigations of idiopathic epilepsy in the Bernese mountain dog. Journal of Small Animal Practice 40, 319–325.
Kearsley-Fleet, L., ONeill, D.G., Volk, H.A., Church, D.B. and Brodbelt, D.C. (2013) Prevalence and risk factors for canine epilepsy of unknown origin in the UK. Veterinary Record 172(13), 338.
Kennerly, E.M., Idaghdour, Y., Olby, N.J., Muñana, K.R. and Gibson, G. (2009) Pharmacogenetic association study of 30 genes with phenobarbital drug response in epileptic dogs. Pharmacogenetics and Genomics 19, 911–922.
Kuwabara, T., Hasegawa, D., Kobayashi, M., Fujita, M. and Orima, H. (2010) Clinical magnetic resonance volumetry of the hippocampus in 58 epileptic dogs. Veterinary Radiology and Ultrasound 51, 485–490.
Kwan, P. and Brodie, M.J. (2000) Early identification of refractory epilepsy. The New England Journal of Medicine 342, 314–319.
Kwan, P., Sills, G.J., Butler, E., Gant, T.W., Meldrum, B.S. and Brodie, M.J. (2002) Regional expression of multidrug resistance genes in genetically epilepsy-prone rat brain after a single audiogenic seizure. Epilepsia 43, 1318–1323.
Kwan, P., Poon, W.S., Ng, H.K., Kang, D.E., Wong, V., Ng, P.W., Lui, C.H., Sin, N.C., Wong, K.S. and Baum, L. (2008) Multidrug resistance in epilepsy and polymorphisms in the voltage-gated sodium channel genes SCN1A, SCN2A, and SCN3A: correlation among phenotype, genotype, and mRNA expression. Pharmacogenetics and Genomics 18, 989–998.
Kwan, P., Arzimanoglou, A., Berg, A.T., Brodie, M.J., Allen Hauser, W., Mathern, G., Moshé, S.L., Perucca, E., Wiebe, S. and French, J. (2010) Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 51, 1069–1077.
Kwan, P., Schachter, S.C. and Brodie, M.J. (2011) Drug-resistant epilepsy. The New England Journal of Medicine 365, 919–926.
Lancaster, E., Lai, M., Peng, X., Hughes, E., Constantinescu, R., Raizer, J., Friedman, D., Skeen, M.B., Grisold, W., Kimura, A., Ohta, K., Iizuka, T., Guzman, M., Graus, F., Moss, S.J., Balice-Gordon, R. and Dalmau, J. (2010) Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurology 9, 67–76.
Löscher, W. and Brandt, C. (2010) High seizure frequency prior to antiepileptic treatment is a predictor of pharmacoresistant epilepsy in a rat model of temporal lobe epilepsy. Epilepsia 51, 89–97.
Löscher, W. and Potschka, H. (2002) Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. The Journal of Pharmacology and Experimental Therapeutics 301, 7–14.
Löscher, W. and Potschka, H. (2005) Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Progress in Neurobiology 76, 22–76.
Masuyama, H., Suwaki, N., Tateishi, Y., Nakatsukasa, H., Segawa, T. and Hiramatsu, Y. (2005) The pregnane X receptor regulates gene expression in a ligand- and promoter-selective fashion. Molecular Endocrinology 19, 1170–1180.
McKnight, K., Jiang, Y., Hart, Y., Cavey, A., Wroe, S., Blank, M., Shoenfeld, Y., Vincent, A., Palace, J. and Lang, B. (2005) Serum antibodies in epilepsy and seizure-associated disorders. Neurology 65, 1730–1736.
Mealey, K.L., Bentjen, S.A., Gay, J.M. and Cantor, G.H. (2001) Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics 11, 727–733.
Miller, D.S. (2010) Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends in Pharmacological Sciences 31, 246–254.
Monteiro, R., Adams, V., Keys, D. and Platt, S.R. (2012) Canine idiopathic epilepsy: prevalence, risk factors and outcome associated with cluster seizures and status epilepticus. Journal of Small Animal Practice 53, 526–533.
Muñana, K.R., Zhang, D. and Patterson, E.E. (2010) Placebo effect in canine epilepsy trials. Journal of Veterinary Internal Medicine 24, 166–170.
Muñana, K.R., Nettifee-Osborne, J.A., Bergman, R.L., Jr and Mealey, K.L. (2012a) Association between ABCB1 genotype and seizure outcome in collies with epilepsy. Journal of Veterinary Internal Medicine 26, 1358–1364.
Pathophysiology of Pharmacoresistant Epilepsy
Muñana, K.R., Thomas, W.B., Inzana, K.D., Nettifee-Osborne, J.A., McLucas, K.J., Olby, N.J., Mariani, C.J. and Early, P.J. (2012b) Evaluation of levetiracetam as adjunctive treatment for refractory canine epilepsy: a randomized, placebo-controlled, crossover trial. Journal of Veterinary Internal Medicine 26, 341–348.
Pakozdy, A., Halasz, P., Klang, A., Bauer, J., Leschnik, M., Tichy, A., Thalhammer, J.G., Lang, B. and Vincent, A. (2013) Suspected Limbic Encephalitis and Seizure in Cats Associated with Voltage-Gated Potassium Channel (VGKC) Complex Antibody. Journal of Veterinary Internal Medicine 27, 212–214.
Pardridge, W.M. (1999) Blood-brain barrier biology and methodology. Journal of Neurovirology 5, 556–569.
Pekcec, A., Unkruer, B., Schlichtiger, J., Soerensen, J., Hartz, A.M., Bauer, B., van Vliet, E.A., Gorter, J.A. and Potschka, H. (2009a) Targeting prostaglandin E2 EP1 receptors prevents seizure-associated P-glycoprotein up-regulation. The Journal of Pharmacology and Experimental Therapeutics 330, 939–947.
Pekcec, A., Unkruer, B., Stein, V., Bankstahl, J.P., Soerensen, J., Tipold, A., Baumgartner, W. and Potschka, H. (2009b) Over-expression of P-glycoprotein in the canine brain following spontaneous status epilepticus. Epilepsy Research 83, 144–151.
Penning, V.A., Connolly, D.J., Gajanayake, I., McMahon, L.A., Luis Fuentes, V., Chandler, K.E. and Volk, H.A. (2009) Seizure-like episodes in 3 cats with intermittent high-grade atrioventricular dysfunction. Journal of Veterinary Internal Medicine 23, 200–205.
Picot, M.C., Baldy-Moulinier, M., Daures, J.P., Dujols, P. and Crespel, A. (2008) The prevalence of epilepsy and pharmacoresistant epilepsy in adults: a population-based study in a Western European country. Epilepsia 49, 1230–1238.
Placencia, M., Sander, J.W., Shorvon, S.D., Roman, M., Alarcon, F., Bimos, C. and Cascante, S. (1993) Antiepileptic drug treatment in a community health care setting in northern Ecuador: a prospective 12-month assessment. Epilepsy Research 14, 237–244.
Platt, S.R., Adams, V., Garosi, L.S., Abramson, C.J., Penderis, J., De Stefani, A. and Matiasek, L. (2006) Treatment with gabapentin of 11 dogs with refractory idiopathic epilepsy. Veterinary Record 159, 881–884.
Podell, M. and Fenner, W.R. (1993) Bromide therapy in refractory canine idiopathic epilepsy. Journal of Veterinary Internal Medicine 7, 318–327.
Potschka, H. (2010) Modulating P-glycoprotein regulation: future perspectives for pharmacoresistant epilepsies? Epilepsia 51, 1333–1347.
Potschka, H. (2012) Role of CNS efflux drug transporters in antiepileptic drug delivery: Overcoming CNS efflux drug transport. Advanced Drug Delivery Reviews 64, 943–952.
Potschka, H., Fedrowitz, M. and Löscher, W. (2002) P-Glycoprotein-mediated efflux of phenobarbital, lamotrigine, and felbamate at the blood-brain barrier: evidence from microdialysis experiments in rats. Neuroscience Letters 327, 173–176.
Potschka, H., Volk, H.A. and Löscher, W. (2004) Pharmacoresistance and expression of multidrug transporter P-glycoprotein in kindled rats. Neuroreport 15, 1657–1661.
Rao, V.V., Dahlheimer, J.L., Bardgett, M.E., Snyder, A.Z., Finch, R.A., Sartorelli, A.C. and Piwnica-Worms, D. (1999) Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proceedings of the National Academy 96, 3900–3905.
Regesta, G. and Tanganelli, P. (1999) Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Research 34, 109–122.
Remy, S., Gabriel, S., Urban, B.W., Dietrich, D., Lehmann, T.N., Elger, C.E., Heinemann, U. and Beck, H. (2003) A novel mechanism underlying drug resistance in chronic epilepsy. Annals of Neurology 53, 469–479.
Sander, J.W. and Shorvon, S.D. (1996) Epidemiology of the epilepsies. Journal of Neurology, Neurosurgery, and Psychiatry 61, 433–443.
Schinkel, A.H., Smit, J.J., van Tellingen, O., Beijnen, J.H., Wagenaar, E., van Deemter, L., Mol, C.A., van der Valk, M.A., Robanus-Maandag, E.C. and te Riele, H.P. (1994) Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77, 491–502.
Schinkel, A.H., Wagenaar, E., Mol, C.A. and van Deemter, L. (1996) P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. Journal of Clinical Investigation 97, 2517–2524.
Schmied, O., Scharf, G., Hilbe, M., Michal, U., Tomsa, K. and Steffen, F. (2008) Magnetic resonance imaging of feline hippocampal necrosis. Veterinary Radiology and Ultrasound 49, 343–349.
Schrickx, J.A. and Fink-Gremmels, J. (2008) Implications of ABC transporters on the disposition of typical veterinary medicinal products. European Journal of Pharmacology 585, 510–519.
Schwartz-Porsche, D. (1986) Epidemiological, clinical and pharmacokinetic studies in spontaneously epileptic dogs and cats. Proceedings of the American College of Veterinary Internal Medicine, pp. 1161–1163.
Schwartz-Porsche, D., Löscher, W. and Frey, H.H. (1985) Therapeutic efficacy of phenobarbital and primidone in canine epilepsy: a comparison. Journal of Veterinary Pharmacology and Therapeutics 8, 113–119.
Seppala, E.H., Jokinen, T.S., Fukata, M., Fukata, Y., Webster, M.T., Karlsson, E.K., Kilpinen, S.K., Steffen, F., Dietschi, E., Leeb, T., Eklund, R., Zhao, X., Rilstone, J.J., Lindblad-Toh, K., Minassian, B.A. and Lohi, H. (2011) LGI2 truncation causes a remitting focal epilepsy in dogs. PLoS Genetics 7, e1002194.
Shihab, N., Bowen, J. and Volk, H.A. (2011) Behavioral changes in dogs associated with the development of idiopathic epilepsy. Epilepsy and Behavior 21, 160–167.
Shukla, S.J., Sakamuru, S., Huang, R., Moeller, T.A., Shinn, P., Vanleer, D., Auld, D.S., Austin, C.P. and Xia, M. (2011) Identification of clinically used drugs that activate pregnane X receptors. Drug Metabolism and Disposition: the Biological Fate of Chemicals 39, 151–159.
Sisodiya, S.M., Lin, W.R., Harding, B.N., Squier, M.V. and Thom, M. (2002) Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain 125, 22–31.
Spector, R. (2000) Drug transport in the mammalian central nervous system: multiple complex systems. A critical analysis and commentary. Pharmacology 60, 58–73.
Tishler, D.M., Weinberg, K.I., Hinton, D.R., Barbaro, N., Annett, G.M. and Raffel, C. (1995) MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia 36, 1–6.
Trepanier, L.A., Van Schoick, A., Schwark, W.S. and Carrillo, J. (1998) Therapeutic serum drug concentrations in epileptic dogs treated with potassium bromide alone or in combination with other anticonvulsants: 122 cases (1992-1996). Journal of the American Veterinary Medical Association 213, 1449–1453.
Vincent, A., Irani, S.R. and Lang, B. (2010) The growing recognition of immunotherapy-responsive seizure disorders with autoantibodies to specific neuronal proteins. Current Opinion in Neurology 23, 144–150.
Volk, H.A. and Löscher, W. (2005) Multidrug resistance in epilepsy: rats with drug-resistant seizures exhibit enhanced brain expression of P-glycoprotein compared with rats with drug-responsive seizures. Brain 128, 1358–1368.
Volk, H.A., Burkhardt, K., Potschka, H., Chen, J., Becker, A. and Löscher, W. (2004a) Neuronal expression of the drug efflux transporter P-glycoprotein in the rat hippocampus after limbic seizures. Neuroscience 123, 751–759.
Volk, H.A., Potschka, H. and Löscher, W. (2004b) Increased expression of the multidrug transporter P-glycoprotein in limbic brain regions after amygdala-kindled seizures in rats. Epilepsy Research 58, 67–79.
Volk, H.A., Arabadzisz, D., Fritschy, J.M., Brandt, C., Bethmann, K. and Löscher, W. (2006) Antiepileptic drug-resistant rats differ from drug-responsive rats in hippocampal neurodegeneration and GABA(A) receptor ligand binding in a model of temporal lobe epilepsy. Neurobiology of Disease 21, 633–646.
Volk, H.A., Matiasek, L.A., Luján Feliu-Pascual, A., Platt, S.R. and Chandler, K.E. (2008) The efficacy and tolerability of levetiracetam in pharmacoresistant epileptic dogs. The Veterinary Journal 176, 310–319.
von Klopmann, T., Rambeck, B. and Tipold, A. (2007) Prospective study of zonisamide therapy for refractory idiopathic epilepsy in dogs. Journal of Small Animal Practice 48, 134–138.
Voss, L.J., Jacobson, G., Sleigh, J.W., Steyn-Ross, A. and Steyn-Ross, M. (2009) Excitatory effects of gap junction blockers on cerebral cortex seizure-like activity in rats and mice. Epilepsia 50(8), 1971–1978.
Waldbaum, S. and Patel, M. (2010) Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Research 88, 23–45.
Weissl, J., Hülsmeyer, V., Brauer, C., Tipold, A., Koskinen, L.L., Kyostila, K., Lohi, H., Sauter-Louis, C., Wolf, M. and Fischer, A. (2012) Disease progression and treatment response of idiopathic epilepsy in Australian Shepherd dogs. Journal of Veterinary Internal Medicine 26, 116–125.
Wessmann, A., Volk, H.A., Parkin, T., Ortega, M. and Anderson, T.J. (2012) Living with canine idiopathic epilepsy: A questionnaire-based evaluation of quality of life. Journal of Veterinary Internal Medicine 26, 1.
3 Classification of Seizures and Epilepsies
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
A seizure (or ictus) has been defined as ’a transient occurrence of signs due to abnormal excessive or synchronous neuronal activity in the brain’ (Fisher et al., 2005). The clinical manifestations of a seizure are sudden and transient and depend on location of onset in the brain, patterns of propagation and a variety of other factors (see Chapter 1). Seizures can affect one or more of the following functions: sensory, motor, and autonomic activity, consciousness, emotional state, memory, cognition or behaviour (Fisher et al., 2005). The seizure (or ictus) may be preceded by a prodrome (or prodromal phase), which can be characterized by anxiety, restlessness, increased affection, withdrawal, aggressiveness, or vocalization, and by an aura, which is the initial manifestation of a seizure. The prodrome can occur hours to days before the seizure, the aura generally lasts seconds. The aura has been described in people as a subjective sensation, such as dizziness, tingling, and anxiety at the start of a seizure. In animals it may manifest as increased or decreased attention seeking, stereotypical sensory or motor behaviour (e.g. licking, pacing) or autonomic manifestations (e.g. salivating, vomiting, urinating). The ictus is the seizure itself and, in most cases, it lasts only a few minutes. The post-ictal period occurs soon after the seizure (or ictus) and may last seconds to days. Clinical manifestations include disorientation, aggressive behaviour, restlessness, pacing, lethargy, deep sleep, hunger, thirst, defecation, urination, ataxia, proprioceptive deficits and decreased or absent menace response with or without actual blindness.
Epilepsy has been defined as an enduring disorder of the brain that is characterized by recurrent seizures (Blume et al., 2001; Fisher et al., 2005). As there are many causes of chronic recurrent seizures, epilepsy is not a specific disease but rather a group of heterogeneous conditions. However, not all seizures are associated with epilepsy. For instance, a seizure can be the reaction of a normal brain to a transient insult, such as intoxication or metabolic disorder. If seizures no longer occur when the metabolic or toxic disorder resolves, the patient is not considered to have epilepsy.
A classification of seizures and epilepsies is important as clinical manifestations and aetiologies of seizures vary considerably. A standardized and uniform classification of seizure and epilepsy would allow consistency in the use of diagnostic terms, improve communication among clinicians and methods of evaluating treatment, and facilitate comparison of clinical cases and scientific studies.
Classification of seizures and epilepsies in veterinary medicine is largely based on its human counterpart and focuses on seizure phenomenology and aetiology (Schwartz-Porsche,
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
1994; Podell et al., 1995; Berendt and Gram, 1999; Licht et al., 2002; Podell, 2004). The main limitations of the veterinary classification are that: (i) recognition of seizure occurrence and clinical manifestations is largely dependent on the pet-owner’s observation; (ii) electroencephalographic (EEG) data are usually not available; and (iii) no agreement has been reached for a standardized terminology in veterinary medicine. Therefore, the veterinary literature on this subject is often confusing with regard to definitions and interpretations. In addition, as in human medicine, seizure classification is an ongoing process and therefore updating is necessary.
This chapter will initially present the definitions and classifications of seizure and epilepsy in human medicine that are relevant in order to understand how terminology and classification have been developed and could evolve in veterinary medicine. Subsequently the focus will be on proposed veterinary terminology and classification.
Classification of Seizures and Epilepsies in Human Medicine
In human medicine, the International League Against Epilepsy (ILAE) has appointed a task force (Commission on Classification and Terminology) for the ongoing process of seizure and epilepsy classification and terminology. Classification systems have been developed based on ictal phenomenology, associated EEG findings and on aetiology. The Commission on Classification and Terminology of the ILAE first published its classification in 1969, which has subsequently been updated in 1981 for seizures (Commission, 1981) and in 1989 for epilepsies (Commission, 1989). Advances in molecular genetics, structural and functional neuroimaging, and neurophysiologic techniques have greatly improved the knowledge and understanding of seizures and epilepsies in humans, creating the need for a more modern system. Attempts have been made to update the 1989 and 1981 classification in 2001 and 2006 (Engel, 2001, 2006), and the most recent proposal for a revised classification has been published in 2010 (Berg et al., 2010) (Table 3.1).
Interestingly, epilepsies of unknown cause account for one-third or more of all epilepsies in humans. It is likely that several epilepsies classified as idiopathic in veterinary medicine (particularly in cats) would be more appropriately classified as unknown (or of unknown aetiology despite extensive investigations) based on the most recent ILAE classification. Future research in neuroimaging and genetics should help to further characterize and better classify these types of epilepsy (Berg et al., 2010).
In human medicine, epilepsies have also been classified as electroclinical syndromes (Berg et al., 2010). An electroclinical syndrome has been defined as a complex of clinical features, signs and symptoms that together define a distinctive, recognizable clinical disorder (Berg et al., 2010). Electroclinical syndromes are distinctive disorders identifiable on the basis of a typical age onset, specific seizure types, EEG characteristics and other factors (such as patterns of seizure occurrence with respect to sleep, provoking or triggering factors), which, when taken together, permit a specific diagnosis. The diagnosis of an electroclinical syndrome often has implications for management, treatment and prognosis (Berg et al., 2010). One of the most distinctive and clinically relevant classifications of the currently recognized electroclinical syndromes in humans is based on typical age at onset (Berg et al., 2010).
The subdivision of generalized seizures into tonic-clonic, tonic, clonic, myoclonic, atonic and absence seizures is overall unchanged since 1981. The 1981 subclassification of absence seizures has been simplified and altered. Absence seizures are characterized by a brief impairment of consciousness and may be associated with mild clonic, tonic, atonic and autonomic manifestations. Absence seizures are subclassified as typical, atypical and with special features (myoclonic absence and eyelid myoclonia) in the latest classification (Berg et al., 2010). Typical absence seizures occur in young patients with idiopathic generalized epilepsies and are usually associated with generalized 3–4 Hz spike-and-slow-wave complexes on EEG. Atypical absence seizures occur in patients with cryptogenic or structural generalized epilepsies and are often associated with slow spike-wave complexes of 1.5–2.5 Hz. Motor manifestations are more pronounced
Table 3.1. Comparison of major changes between the 1981 and 1989 Classification and Terminology and the newly proposed Classification and Terminology (Commission, 1981, 1989; Berg et al., 2010).
ILAE 1981 and 1989 ILAE 2010
Generalized seizures are those in which the first clinical and electroencephalographic changes indicate initial involvement of both cerebral hemispheres.
Focal seizures (previously termed as partial) are those in which the first clinical and electroencephalographic changes indicate initial activation of a system of neurons limited to a part of one cerebral hemisphere.
Idiopathic epilepsy: there is no underlying cause other than a possible hereditary predisposition. Idiopathic epilepsies are defined by age-related onset, clinical and electroencephalographic characteristics, and a presumed genetic aetiology.
Structural epilepsy: this type of epilepsy is the consequence of a known or suspected disorder of the central nervous system.
Cryptogenic (probable structural) epilepsy: this refers to a disorder whose cause is hidden or occult. Cryptogenic epilepsies are presumed to be structural.
Generalized seizures are conceptualized as originating at some point within and rapidly engaging bilaterally distributed networks. Such bilateral networks can include cortical and subcortical structures, but do not necessarily include the entire cortex.
Focal seizures are conceptualized as originating at some point within networks limited to one hemisphere. They may be discretely localized or more widely distributed. Focal seizures may originate in subcortical structures.
Genetic epilepsy: the epilepsy is, as best as understood, the direct result of a known or presumed genetic defect(s) in which seizures are the core symptom of the disorder. This attribution must be supported by specific forms of evidence (e.g. specific molecular genetic studies or family studies).
Structural and metabolic epilepsy: this type of epilepsy is the secondary result of a distinct structural or metabolic condition. These structural or metabolic disorders may be of acquired or genetic origin (as is the case for malformations of cortical development and certain metabolic disorders).
Unknown epilepsy: the nature of the underlying cause is as yet unknown; it may have a fundamental genetic basis (e.g. a previously unrecognized channelopathy) or it may be the consequence of an unrecognized structural or metabolic disorder not yet identified.
in patients with atypical absence seizures than in those with typical absence seizures.
The 1981 classification subdivided focal seizure types into: complex (with impairment of consciousness), simple (without impairment of consciousness) and secondarily generalized (when the initially focal ictal event progresses to involvement of the entire body resulting in a generalized seizure) (Commission, 1981). The most recently proposed classification (Berg et al., 2010) recommends to abandon this terminology (simple, complex, secondarily generalized) and to describe focal seizure semiology accurately, including impairment of consciousness/awareness, when recognized, and localization and progression of ictal events.
The most recently proposed classification of seizure and epilepsies (Berg et al., 2010) has been met with considerable dissatisfaction from several expert epileptologists and therefore the previous ILAE classifications are likely to continue to be used until a more widely accepted proposal has been developed (Panayiotopoulos, 2011, 2012). Among the criticisms to the ILAE 2010 proposal (Berg et al., 2010) the comments that are most relevant to the current veterinary classification involve:
although their correct definition should be reiterated. The term ‘genetic’ should be introduced, not to replace ‘idiopathic’ but to represent a new category in addition to the other three in a revised classification of epilepsies (Panayiotopoulos, 2012).
Classification of Seizures and Epilepsies in Veterinary Medicine
The following classification of canine and feline seizures (Tables 3.2 and 3.3) is largely based on the ILAE classifications (mainly Commission, 1981, 1989; Engel, 2001, 2006) and on selected veterinary literature (Schwartz-Porsche, 1994; Berendt and Gram, 1999; Licht et al., 2002; Berendt, 2004; Podell, 2004; Thomas, 2010). The parallelism with the ILAE 2010 proposed new terminology (Berg et al., 2010) is indicated in parentheses where appropriate in Table 3.2, in a specific column in Table 3.3 and also discussed in the text.
Veterinary classification of seizures based on clinical manifestations
Generalized-onset seizures
The initial clinical manifestations of generalized-onset seizures indicate more than minimal involvement of both cerebral hemispheres. The motor manifestations begin bilaterally and are often symmetrical. An alteration in consciousness frequently occurs at some stage during ictus. The term ‘primary generalized’ has sometimes been used in the veterinary literature to describe generalized-onset seizures and to differentiate them from secondarily generalized focal (partial) seizures. In this case the term primary does not refer to seizure aetiology, but to the fact that the initial clinical manifestations reflect involvement of both cerebral hemispheres simultaneously from the onset of the seizure.
generalised-onset tonic-clonic seizure. The most common type of generalized-onset seizure in dogs is the tonic-clonic seizure (formerly called grand mal seizure) (Berendt and Gram, 1999; Licht et al., 2002). A prodromal phase and aura are not always recognized by the owner. Their duration and clinical manifestations are variable. The prodromal phase can last hours to days and it is commonly characterized by restlessness, anxiety or reluctance to perform normal activities. The aura may manifest as increased or decreased attention seeking, stereotypical sensory or motor behaviour (e.g. licking, pacing) or autonomic manifestations (e.g. salivating, vomiting, urinating). The ictal phase of a tonic-clonic seizure is characterized by sustained contraction of all muscles resulting in rigid extension of the limbs and opisthotonos, usually lasting 10 to 60 s (tonic phase). The animal falls on its side and often loses consciousness (defined as attentiveness or responsiveness to the owner and other external stimuli). Breathing is often irregular or absent, and
Table 3.2. Seizure classification based on clinical manifestations.
Generalized-onset seizures Tonic-clonic Tonic Clonic Myoclonic Atonic Absence
Focal-onset seizures Motor Autonomic Sensory Simple (without impairment of
consciousness/awareness) Complex (with impairment of consciousness/awareness) Secondarily generalized (evolving to a bilateral tonic,
clonic or tonic-clonic seizure)
cyanosis is common. The tonic phase is followed by the clonic phase, which is characterized by rhythmic muscular contractions resulting in uncoordinated, purposeless, jerking movements of the limbs and chewing movements. Automatisms such as running or paddling movements of the limbs commonly occur. The clonic phase may alternate with tonic activity. Autonomic manifestations, such as hypersalivation, urination, defecation and mydriasis are common in dogs with generalized-onset tonic-clonic seizures although not a constant feature. The ictus usually lasts 1 to 2 min. The post-ictal phase duration and clinical manifestations are variable. The dog may rest and rapidly return to normal activity or may show confusion, disorientation, aggressive behaviour, restlessness, pacing, lethargy, deep sleep, hunger, thirst, defecation, urination, ataxia, proprioceptive deficits, and decreased or absent menace response with or without actual blindness, which can persist for 24 h or longer. The post-ictal phase duration and severity of clinical manifestations may be unrelated to the duration and severity of the ictus.
Tonic-clonic seizures also have been reported as the most common type of generalized-onset seizures in cats (Schwartz-Porsche and Kaiser, 1989; Schriefl et al., 2008). During the pre-ictal phase cats may display aggressiveness (hissing and growling), vocalization (including growling and crying), restlessness (running around erratically), anxiety, hiding or increased affection (seeking refuge with the owner), or may act as fearful. The clinical manifestations of the ictus can be violent and sometimes result in self-inflicted trauma such as excoriations, contusions, avulsion of nails and biting of the tongue (Schwartz-Porsche and Kaiser, 1989; Quesnel et al., 1997). Cats may be propelled forward, up in the air and from side to side. Facial muscle twitching is often observed before or after the tonic phase of the seizure. Autonomic manifestations such as mydriasis, salivation, piloerection and urination and occasionally defecation can also occur (Quesnel et al., 1997). The ictal phase usually lasts 30 s to 2 min. The postictal phase is similar to dogs, lasting minutes to several hours or days and is characterized by restlessness, disorientation, aimless wandering, thirst and hunger. Both cats and dogs may sleep for a few hours after the termination of the seizure.
generalized-onset tonic seizures. Tonic seizures are characterized by sustained increase in muscle contraction without clonic motor activity. Impairment of consciousness and autonomic manifestations can occur. The pre- and postictal phases are similar to those described for generalized-onset tonic-clonic seizure.
generalized-onset clonic seizures. Clonic seizures are characterized by regularly repetitive, sudden, brief, involuntary contractions, which involve the same muscle groups, and are prolonged. Impairment of consciousness and autonomic manifestations can occur. The pre- and post-ictal phases are similar to those described for generalized-onset tonic-clonic seizure.
Tonic or clonic seizures alone have been reported uncommonly in dogs and cats (Schwartz-Porsche and Kaiser, 1989; Heynold et al., 1997; Licht et al., 2002, 2007).
generalized-onsetmyoclonic seizures. Myoclonic seizures are characterized by sudden, brief, involuntary, shock-like contractions that can be generalized or confined to individual muscles or muscle groups (e.g. face, trunk, one extremity).
This type of seizure has been reported in the miniature wire-haired dachshund, beagle and basset hound dogs in association with Lafora disease (Jian et al., 1990; Fitzmaurice et al., 2001; Gredal et al., 2003). The clinical presentation is characterized by repetitive, brief myoclonic jerking of the head, neck and thoracic limbs that are frequently strong enough to cause the animal to fall backward into a sitting or lying position (Davis et al., 1990; Fitzmaurice et al., 2001; Schoeman et al., 2002). Myoclonic seizures may occur spontaneously or in response to visual (including light), tactile or auditory stimuli (Fitzmaurice et al., 2001; Webb et al., 2009). Myoclonic seizures have been reported also in cats (Schwartz-Porsche, 1989). Not all myoclonic jerks are seizures, as they can result from other causes.
generalized-onset atonic seizures. Atonic seizures are characterized by sudden loss of postural
L. De Risio
tone of the head, one limb, or of the entire body usually lasting 1, 2 or more seconds. Consciousness may be lost. This type of seizure has been reported in dogs (Podell, 2004) and needs to be differentiated from narcolepsy/cataplexy and syncope (see Table 9.1, Chapter 9). Generalized seizures characterized by collapse, loss of consciousness and minimal limb movements have been reported in a cat with structural epilepsy (Barnes et al., 2004) and may represent a form of atonic seizure activity.
generalized-onset absence seizures. Absence seizures (formerly called petit mal seizures) are characterized by a transient and brief impairment of consciousness associated with a characteristic EEG pattern (2.5- to 4-Hz spike-and-wave complexes) (see Fig. 11.12, Chapter 11). Absence seizures have been clinically described as a transient cessation of activity with staring, unresponsiveness and ‘blanking out’ episodes. Absence seizures with myoclonic features have been reported in an 8-month-old male Chihuahua that presented with recurrent episodes of head and nose twitching associated with intermittent hind-limb jerking and suspected staring for a few seconds (Poma et al., 2010). Video-EEG documented multiple staring episodes either alone or in association with head and/or nose myoclonic jerks associated with generalized bilaterally synchronous 4 Hz spike-and-wave complexes (Poma et al., 2010). Absence seizures may occur also in cats, but they have not been reported as such or supported by EEG findings. One feline study reported a ‘mild’ form of generalized seizure activity characterized by transient cessation of activity, impaired consciousness, and bilateral facial muscle twitching for a few to several seconds (Quesnel et al., 1997). These may have been a form of absence seizures in cats. Unless absence seizures are frequent, associated with some motor manifestations or the pet-owner is very observant, they go unrecognized. Video-EEG monitoring is very helpful in the diagnosis of this type of seizure.
Focal-onset seizures
The clinical manifestations of focal-onset seizures (formerly called partial seizures) indicate initial abnormal neuronal activity localized in a particular area of a cerebral hemisphere (seizure focus). The newly proposed ILAE classification (Berg et al., 2010) introduces the concept of the seizure focus being discretely localized or more widely distributed within the affected hemisphere.
The clinical manifestations can vary considerably depending on the function of the affected cerebral area and include involuntary motor activity, autonomic signs, sensory abnormalities, alterations of consciousness and paroxysms of abnormal behaviour. These clinical manifestations may occur alone or in various combinations.
focal-onset motor seizures. Focal-onset motor seizures are characterized by involuntary, generally unilateral motor activity resulting in abnormal movements of a body part, such as turning the head to one side, flexion and/or extension of one limb, contraction of facial or masticatory muscles. Focal-onset motor seizures are presumed to arise from a seizure focus near a primary motor area in the frontal cortex contralateral to the observed involuntary motor activity.
focal-onset autonomic seizures. Focal-onset autonomic seizures result in one or more autonomic manifestation including mydriasis, hypersalivation, piloerection, lacrimation, urination, defecation, vomiting, diarrhoea and apparent abdominal pain (Breitschwerdt et al., 1979; Licht et al., 2002; Berendt et al., 2004). Phenobarbitone-responsive hypersalivation, dysphagia, salivary gland enlargement and oesophageal spasms have been reported in a few dogs and may be a form of focal autonomic seizure (Stonehewer et al., 2000; Gibbon et al., 2004).
focal-onset sensory seizures. Focal-onset sensory seizures result in abnormal sensations such as paraesthesia (numbness, tingling) limited to a defined somatosensory region of the body, or in visual hallucinations. Sensory seizures have been subclassified into somatosensory and special sensory (visual, auditory, olfactory, gustatory and vestibular) in humans (Commission, 1981). It is likely that the same sensory disturbances occur in animals, but they are difficult or impossible to identify and associate with concurrent EEG abnormalities. Therefore it can only be speculated that episodes characterized by chewing and/or licking into the air or at a specific region of the body, rubbing of the face, or biting at imaginary objects (‘fly biting’ or ‘fly catching’) are the manifestation of sensory seizures. Repetitive episodes of ‘fly biting’ could be the consequence of focal sensory seizures in the visual cortex, similar to focal visual sensory seizures that occur in humans (Cash and Blauch, 1979; Licht et al., 2002), could result from focal seizures with paroxysms of abnormal behaviour (Berendt et al., 2004), or may represent a form of compulsive behavioural disorder (see Chapter 9) (Rusbridge, 2005).
In analogy with the human classification published by the ILAE in 1981 and the veterinary medical literature, focal seizures have been referred to as complex and simple depending on whether or not consciousness is altered. However, in the context of the ILAE classification, consciousness was defined as ‘the degree of awareness and/or responsiveness of the patient to externally applied stimuli’. Responsiveness was defined as ‘the ability of the patient to carry out simple commands or willed movement’ and awareness referred ‘to the patient’s contact with events during the period in question and its recall’. These functions are difficult if not impossible to assess in veterinary patients, especially based on the pet-owner description or video documentation (Berendt et al., 2004). Therefore veterinary medicine can follow the most recent recommendation of the ILAE (Berg et al., 2010) to avoid using ictal impairment of consciousness to classify specific seizure types (e.g. simple and complex focal seizures) and to describe individual seizure phenomenology accurately including impairment of consciousness/awareness, when recognized.
Focal seizures can also occur as stereotyped paroxysms of abnormal behaviour including attention-seeking or avoidance/escaping behaviour, aimless wandering, restlessness and unprovoked aggression (Licht et al., 2002; Berendt et al., 2004). Paroxysmal abnormal behaviour may result from a manifestation of sensory seizures, involvement of the limbic system or higher cerebral activity (psychic seizures according to the 1981 ILAE classification). Episodes characterized by impaired consciousness (‘trance-like staring’), abnormal behaviour, including unprovoked aggression, extreme irrational fear, compulsive tail-chasing and fly catching, have been reported as focal seizures in eight bull terriers with interictal EEG abnormalities (multiple epileptiform spikes) and moderate to severe ventriculomegaly on computed tomography (Dodman et al., 1996).
The terms complex partial (or focal) seizures, psychomotor seizures, temporal lobe and limbic seizures or epilepsy have been used interchangeably in the veterinary literature to indicate focal seizures characterized by paroxysms of abnormal behaviour with or without some degree of impairment of consciousness.
Any type of focal-onset seizure can evolve into a generalized seizure (secondarily generalize).
The onset of ictus is characterized by clinical (usually motor) manifestations consistent with the location of the seizure focus and within seconds to minutes seizure activity spreads to involve both cerebral hemispheres resulting in bilaterally symmetrical motor disturbances (usually tonic-clonic), autonomic dysfunction and (commonly) altered consciousness. The focal onset may be subtle and the secondary generalization can occur so rapidly that the initial focal component is undetected and the seizure is misclassified as a generalized-onset seizure. Close observation is essential to recognize the focal-onset of the seizure before its secondary generalization. If the terminology recently proposed by the ILAE (Berg et al., 2010) is embraced also in veterinary medicine, the term ‘secondarily generalized’ should be abandoned and replaced by a description of localization and progression of ictal events.
Focal-onset seizures have also been reported in cats (Quesnel et al., 1997; Barnes et al., 2004; Schriefl et al., 2008; Pákozdy et al., 2010). As in dogs, clinical manifestations include motor, sensory and autonomic signs, alterations of consciousness, and paroxysms of abnormal behaviour, which can occur alone or in various combinations. Reported clinical manifestations include lack of response to
L. De Risio
sensory stimuli, unilateral facial twitching (that can be limited to the ear, lip, or eyelids), turning the head to one side, repetitive movements of one or both limbs on one side of the body, mydriasis, hypersalivation, urination, abnormal behaviour suggestive of some form of hallucinations (unjustified hissing, growling, piloerection, attacking a real or imaginary object, unprovoked startle, running frantically, often blindly, into objects), self-chewing, biting and circling (Schwartz-Porsche and Kaiser, 1989; Quesnel et al., 1997). As in dogs, focal seizures have been reported to secondarily generalize in cats (Quesnel et al., 1997; Schriefl et al., 2008).
A peculiar type of feline focal seizures characterized by orofacial involvement, impaired consciousness, occurrence in clusters and frequent association with hippocampal pathology has been described (Pákozdy et al., 2011). Ictal signs include hypersalivation, facial twitching, lip smacking, chewing, licking, swallowing, mydriasis, motionless staring and vocalization. Secondary generalization occurs in the majority (12/15, 70%) of cats. The most common post-ictal and interictal signs are behavioural changes and aggression. Antibodies against voltage-gated potassium channel complexes may play a role in the pathogenesis of this type of feline focal seizure (Pakozdy et al., 2013).
In addition to a classification based on clinical manifestations, seizures have also been classified based on underlying aetiology (Table 3.3). The aetiology-based classification allows for a more specific differential diagnosis. Reported aetiologies of reactive and structural seizures in dogs and cats are described in detail in Chapters 4 and 5, respectively. Idiopathic epilepsy is presented in Chapter 6.
Veterinary classification of seizures and epilepsies based on underlying aetiology
Reactive seizures
Reactive seizures are the reaction of a normal brain to a systemic metabolic or nutritional disorder or exogenous toxin exposure (Podell et al., 1995). When the metabolic, nutritional or toxic disorder resolves, the animal does not have recurrent seizures, and therefore reactive seizures are not considered a form of epilepsy, per se (Jull et al., 2011). The authors feel that the term reactive seizure should continue to be used in veterinary medicine as the term ‘metabolic’ proposed by the ILAE 2010 may generate confusion by referring to metabolic disorders only leaving toxic and nutritional aetiologies poorly classified. In addition the term ‘metabolic’ has not been widely accepted by several expert epileptologists (Panayiotopoulos, 2012).
Animals with reactive seizures frequently have acute onset of bilateral symmetrical neurological deficits indicating diffuse forebrain or intracranial involvement and concurrent signs of dysfunction of other body systems. Occasionally, seizures are the only obvious clinical abnormality. Aetiologies of reactive seizures are listed in Box 3.1 and described in detail in Chapter 4.
Structural (symptomatic or secondary) epilepsy
The terms symptomatic epilepsy and secondary epilepsy have both been used to indicate recurrent seizures caused by a known and identifiable structural forebrain disorder such as vascular, inflammatory/infectious, traumatic, anomalous/developmental, neoplastic and degenerative diseases. The term structural epilepsy, introduced by the ILAE 2010 proposal, should be adopted also in veterinary medicine as its meaning indicates more clearly epilepsy resulting from structural forebrain disease. The term secondary is too generic and prone to misuse, indeed, it has sometimes been used in the veterinary literature to refer to seizures caused by structural forebrain disorders as well as metabolic or toxic disorders (Pákozdy et al., 2010) generating confusion when attempting study comparison. The term symptomatic is a truism as recurrent seizures (epilepsy) are the symptom in humans and the clinical sign in animals of an underlying disease.
Dogs and cats with structural epilepsy usually present with neurological signs (other than seizures) interictally. However, focal lesions in particular areas of the brain (‘clinically silent regions’), such as olfactory bulb and frontal lobes, can result in seizure activity
Table 3.3. Veterinary classification of seizures and epilepsies based on underlying aetiology, parallelism with the ILAE 2010 proposed new terminology, and terminology used in this book.
ILAE 2010 proposed new Current veterinary terminology terminology Terminology used in this book
Reactive seizures Metabolic Reactive seizures
Symptomatic or secondary Structural epilepsy Structural epilepsy epilepsy
Probable symptomatic Epilepsy of unknown aetiology Cryptogenic epilepsy or cryptogenic epilepsy
Idiopathic or Genetic Idiopathic epilepsy primary epilepsy
Box 3.1. Aetiologies of reactive seizures.
Metabolic
Hepatic disease congenital and acquired portosystemic shunt, microvascular dysplasia, hepatic lipidosis, neoplasia, inflammation
Renal disease acute renal failure, end-stage chronic renal failure Electrolyte imbalance hypo- or hypernatraemia, hypocalcemia Hypoglycaemia
insulin-secreting tumour, severe sepsis, iatrogenic insulin overdose Hypoxia Hypertension Polycythaemia
Nutritional
Thiamine deficiency Toxicity Pyrethrins/ Pyrethroids, organophosphates, chlorinated hydrocarbons carbamate Metaldehyde Strychnine, bromathalin Sodium monofluoroacetate (compound 1080) Ethylene glycol Detergents and disinfectants Lead and other heavy metals Poisonous plants Mycotoxins (penitrem A, roquefortine) Animal-related poisoning (toad, spider, bee and wasp venom) Metronidazole (cats) 5-hydroxytryptophan Caffeine and other methylxanthines Amphetamine and amphetamine-like compounds Selective serotonin reuptake inhibitors
without any other neurological signs (Foster et al., 1988; Smith et al., 1989). Aetiologies of structural epilepsy are listed in Box 3.2 and described in detail in Chapter 5.
Classification of certain disorders may be open to debate. For example, organic acidurias such as L-2-hydroxyglutaric aciduria in Staffordshire bull terriers may be classified as structural epilepsy as they result in MRI and histological changes in the brain as well as metabolic disorders as they are caused by an error of cellular metabolism, or genetic epilepsy, as the underlying genetic mutation is known (Abramson et al., 2003; Penderis et al., 2007).
L. De Risio
Box 3.2. Aetiologies of structural epilepsy.
Vascular
Cerebrovascular disease (ischaemic, haemorrhagic) Inflammatory/ infectious
Viral
Bacterial
Rickettsial
Protozoal
Mycotic
Parasitic
Mycoplasmosis
Algal
Granulomatous meningoencephalomyelitis
Necrotizing meningoencephalitis
Necrotizing leukoencephalitis Other feline and canine meningoencephalitis or meningoencephalomyelitis of unknown aetiology Traumatic brain injury Anomalous and developmental
Hydrocephalus
Hydranencephaly
Porencephaly
Meningoencephalocele
Meningoencele
Exencephaly
Holoprosencephaly
Agenesis of the corpus callosum
Lissencephaly
Polymicrogyria
Cerebral neuronal heterotopias or dysplasias Neoplastic
Primary
Meningioma
Astrocytoma (glioblastoma multiforme)
Oligodendroglioma
Gliomatosis cerebri
Ependymoma
Choroid plexus tumours
Primitive neuroectodermal tumours (neuroblastomas, medulloblastoma, gangliocytomas)
Primary CNS lymphomas
Primary CNS histiocytic sarcoma (malignant histiocytosis)
Secondary
Haemangiosarcoma
Lymphoma
Pituitary Tumours
Carcinomas/ Adenocarcinomas (mammary, prostatic, pancreatic, pulmonary)
Nasal tumours (e.g. adenocarcinoma, squamous cell carcinoma, chondrosarcoma,
neuroesthesioblastoma)
Histiocytic sarcoma
Calvarial osteosarcoma and multilobulated tumour of bone (multilobulated osteochondrosarcoma)
Malignant melanoma
Others Degenerative
Lysosomal storage diseases
Organic acidurias
Continued
Box 3.2. Continued.
Mitochondrial encephalopathies and encephalomyelopathies Leukodystrophies Spongy degenerations Multisystem neuronal degeneration/abiotrophy
Probable symptomatic or cryptogenic epilepsy
Probable symptomatic or cryptogenic epilepsy refers to recurrent seizures caused by an underlying brain disease that is strongly suspected but cannot be identified despite extensive investigations (e.g. undetected hypoxic or vascular events, post-encephalitic changes and post-traumatic lesions that do not cause any detectable changes on brain imaging). Animals with cryptogenic epilepsy may or may not have behavioural and/or neurological abnormalities (other than seizures) interictally. The term ‘epilepsy of unknown aetiology’ has been proposed to replace the term cryptogenic epilepsy in humans (Berg et al., 2010) and although not widely accepted by expert epileptologists (Panayiotopoulos, 2012), it may be of value in veterinary medicine due to its more immediate meaning. However, in veterinary medicine, it would be important to specify when the cause of the seizures is unknown despite extensive diagnostic investigations to rule out toxic, metabolic, nutritional and structural brain disorders (see Chapter 10) rather than unknown because of minimal or no investigations.
Idiopathic or primary epilepsy
Idiopathic or primary epilepsy refers to recurrent seizures with no underlying cause other than a strongly suspected or confirmed genetic or familial basis. In this context, the term ‘idiopathic’ refers to a disorder ‘by itself’ not ‘of unknown cause’. The 1989 ILAE classification defined idiopathic epilepsies based on age at seizure onset, clinical and electroencephalographic characteristics, and a presumed genetic aetiology. The diagnosis of idiopathic epilepsy in dogs and cats is based on the age at onset (generally between 6 months and 6 years), normal interictal behaviour, physical and neurological examinations, and exclusion of metabolic, toxic and structural cerebral disorders by means of diagnostic investigations (see Chapter 6). An inherited basis, familial transmission, or a higher incidence of idiopathic epilepsy has been reported in several canine breeds (Table 6.1, Chapter 6). A genetic basis for recurrent seizures has been reported in a closed colony of laboratory cats (Kuwabara et al., 2010). The breed of these cats was not specified. The age at the time of the first seizure ranged between 4 and 12 months. General physical and neurological examinations and results of diagnostic investigations (including 1.5 Tesla MRI of the brain and CSF analysis) were all normal. All cats had focal-onset complex seizures followed by secondary generalization into tonic-clonic seizures. Based on pedigree analysis an autosomal recessive mode of inheritance was hypothesized. In the clinical setting, it is difficult to demonstrate a genetic or familial basis for recurrent seizures, particularly in cats. It is likely that several cats and some of the dogs reported to have idiopathic epilepsy in the veterinary literature actually would be better classified as having epilepsy of unknown aetiology based on the most recent ILAE proposal (Berg et al., 2010) as a genetic or familial basis was neither suspected nor confirmed in these individuals and the diagnostic investigations were not always complete. Applying the term genetic rather than idiopathic also in veterinary medicine, in analogy with the most recent ILEA proposal (Berg et al., 2010), would have the advantage of limiting its use to purebred dogs with a strongly suspected or proven genetic or familial basis for the recurrent seizures. However, this term may generate confusion as clinicians may tend to use it for purebred
L. De Risio
dogs with the typical clinical features of idiopathic epilepsy and suspected genetic aetiology as well as dogs with known genetic mutations (e.g. Epm2b gene in miniature wirehaired dachshunds with autosomal recessive progressive myoclonic epilepsy (Lafora disease), or Lgi2 gene in Lagotto Romagnolo with benign familial juvenile epilepsy) resulting in particular types of epilepsy that do not meet the typical criteria for idiopathic epilepsy (Lohi et al., 2005; Seppala et al., 2011). Therefore it may be more appropriate to continue to use the term idiopathic to indicate dogs with clinical and diagnostic features typical for idiopathic epilepsy as well as a strongly suspected or proven genetic or familial basis for the recurrent seizures. The term genetic epilepsy could be introduced as an additional category to include disorders with known genetic mutation, as recently proposed in humans (Panayiotopoulos, 2012).
While it was originally thought that focal-onset seizures with or without secondary generalization would occur only in animals with structural brain diseases (symptomatic epilepsy), focal-onset seizures have been reported also in dogs and cats with idiopathic epilepsy (Patterson et al., 2003; Berendt et al., 2009; Kuwabara et al., 2010; Pákozdy et al., 2010). In addition, the same animal can be affected by different types of seizures (e.g. focal-onset with or without secondary generalization and generalized-onset seizures) (Quesnel et al., 1997; Licht et al., 2002; Pákozdy et al., 2010). Therefore the clinical manifestations of seizures should not be used to infer the aetiologic diagnosis.
Precipitated seizures
The majority of seizures appear to occur spontaneously, however sometimes seizures may be precipitated by a variety of environmental and internal factors. In human patients, sleep deprivation, emotional stress, menstruation, missed anti-epileptic medication and concurrent illness can result in so called ‘precipitated seizures’ (Commission, 1989). Emotional stress caused by changes in the daily routine (including moving to another place or travelling), unexpected noise, sudden awakening, or an unusual event have been reported to precipitate seizures in Labrador retrievers (Heynold et al., 1997). Another study reported that anxiety, hyperactivity or stress (e.g. working under conditions with a demand for high performance) sometimes provoked seizures in 22% (11/49) of the included Belgian shepherds (Berendt et al., 2008).
Reflex seizures
Reflex seizures are seizures that can be consistently provoked by specific sensations or perceptions (Commission, 1989). The trigger is specific and the latency between trigger and seizure is short (seconds to minutes). Reflex seizure triggers in people include flickering light (usually from a television) or other visual stimuli, immersion in hot water, reading, certain sounds and eating. These stimuli are usually limited in an individual patient to a single specific stimulus or a limited number of closely related stimuli. Reflex seizures are usually generalized (although focal seizures have also been reported in association with tactile or proprioceptive stimuli) and associated with idiopathic epilepsy in humans (Commission, 1989). Seizures consistently associated with sounds (lawnmower engine), automobile rides or veterinary offices have been observed in dogs (Thomas, 2010).
Regardless of the underlying aetiology, seizures can occur as:
Status epilepticus and cluster seizures are neurological emergencies described in detail in Chapters 23 and 24.
Classification of seizures and epilepsies is an ongoing process in humans and veterinary neurologists should follow its development closely. Establishing a universally accepted and standardized terminology to describe ictal phenomenology would greatly help communication among veterinary clinicians and scientists and represent the foundation of further development of the veterinary classification. The veterinary classification of seizures and epilepsies will evolve as EEG and functional MRI become more widely used, new underlying aetiologies are detected, and breed-related epileptic syndromes with specific genetic mutations are identified.
References
Abramson, C.J., Platt, S.R., Jakobs, C. et al. (2003) L-2-Hydroxyglutaric aciduria in Staffordshire bull terriers. Journal of Veterinary Internal Medicine 17, 551–556.
Barnes, H.L., Chrisman, C.L., Mariani, C.L., Sims, M. and Alleman, A.R. (2004) Clinical signs, underlying cause, and outcome in cats with seizures: 17 cases (1997-2002). Journal of American Veterinary Medical Association 225, 1723–1726.
Berendt, M. (2004) Epilepsy. In: Vite, C.H. (ed.) Braund’s Clinical Neurology in Small Animals: localization, diagnosis and treatment. Document No. A3230.0704. International Veterinary Information Service, Ithaca, New York.
Berendt, M. and Gram, L. (1999) Epilepsy and seizure classification in 63 dogs: a reappraisal of veterinary epilepsy terminology. Journal of Veterinary Internal Medicine 13, 14–20.
Berendt, M., Gredal, H. and Alving, J. (2004) Characteristics and phenomenology of epileptic partial seizures in dogs: similarities with human seizure semiology. Epilepsy Research 61, 167–173.
Berendt, M., Gullov, C.H., Christensen, S.L., Gudmundsdottir, H., Gredal, H., Fredholm, M. and Alban, L. (2008) Prevalence and characteristics of epilepsy in the Belgian shepherd variants Groenendael and Tervuren born in Denmark 1995-2004. Acta Veterinaria Scandinava 50, 51.
Berendt, M., Gullov, C.H. and Fredholm, M. (2009) Focal epilepsy in the Belgian shepherd: evidence for simple Mendelian inheritance. Journal of Small Animal Practice 50, 655–661.
Berg, A.T., Berkovic, S.F., Brodie, M.J., Buchhalter, J., Cross, J.H., van Emde Boas, W., Engel, J., French, J., Glauser, T.A., Mathern, G.W., Moshé, S.L., Nordli, D., Plouin, P. and Scheffer, I.E. (2010) Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 51, 676–685.
Blume, W.T., Luders, H.O., Mizrahi, E., Tassinari, C., van Emde Boas, W. and Engel, J. (2001) Glossary of ictal semiology. Epilepsia 42, 1212–1218.
Breitschwerdt, E.B., Breazile, J.E. and Broadhurst, J.J. (1979) Clinical and electroencephalographic findings associated with ten cases of suspected limbic epilepsy in the dog. Journal of the American Animal Hospital Association 15, 37–50.
Cash, W.C. and Blauch, B.S. (1979) Jaw snapping syndrome in eight dogs. Journal of the American Veterinary Medical Association 175, 709–710.
Commission on Classification and Terminology of the International League Against Epilepsy (1981) Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 22, 489–501.
Commission on Classification and Terminology of the International League Against Epilepsy (1989) Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 30, 389–399.
Davis, K.E., Finnie, J.W. and Hooper, P.T. (1990) Lafora’s disease in a dog. Australian Veterinary Journal 67, 192–193.
Dodman, N.H., Knowles, K.E., Shuster, L., Moon-Fanelli, A.A., Tidwell, A.S. and Keen, C.L. (1996) Behavioral changes associated with suspected complex partial seizures in bull terriers. Journal of the American Veterinary Medical Association 208, 688–691.
Engel, J. (2001) A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 42, 796–803.
Engel, J. (2006) Report of the ILAE Classification Core Group. Epilepsia 47, 1558–1568.
Fisher, R.S., van Emde Boas, W., Blume, W., Elger, C., Genton, P., Lee, P. and Engel, J. Jr (2005) Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46, 470–472.
Fitzmaurice, S., Rusbridge, C., Shelton, G.D. et al. (2001) Familial myoclonic epilepsy in the miniature wirehaired dachshund. In: Proceedings of the 14th Annual Symposium, ESVN 2000, p. 29.
Foster, E.S., Carrillo, J.M. and Patnaik, A. (1988) Clinical signs of tumours affecting the rostral cerebrum in 43 dogs. Journal of Veterinary Internal Medicine 2, 71–74.
Gibbon, K.J., Trepanier, L.A. and Delaney, F.A. (2004) Phenobarbital-Responsive ptyalism, dysphagia, and apparent esophageal spasm in a German shepherd puppy. Journal of the American Animal Hospital Association 40, 230–237.
Gredal, H., Berendt, M. and Leifsson, P.S. (2003) Progressive myoclonus epilepsy in a beagle. Journal of Small Animal Practice 44, 511–514.
Heynold, Y., Faissler, D., Steffen, F. and Jaggy, A. (1997) Clinical, epidemiological and treatment results of idiopathic epilepsy in 54 Labrador retrievers: a long-term study. Journal of Small Animal Practice 38, 7–14.
Jian, Z., Alley, M.R., Cayzer, J. and Swinney, G.R. (1990) Lafora’s disease in an epileptic basset hound. New Zealand Veterinary Journal 38, 75–79.
Jull, P., De Risio, L., Horton, C. and Volk, H.A. (2011) Effect of prolonged status epilepticus as a result of intoxication on epileptogenesis in a UK canine population. Veterinary Record 169, 361.
Kuwabara, T., Hasegawaa, D., Ogawa, F., Kobayashia, M., Fujita, M., Suzuki, H., Matsukic, N. and Orima, H. (2010) A familial spontaneous epileptic feline strain: A novel model of idiopathic/genetic epilepsy. Epilepsy Research 92, 85–88.
Licht, B.G., Licht, M.H., Harper, K.M., Lin, S., Curtin, J.J., Hyson, L.L. and Willard, K. (2002) Clinical presentations of naturally occurring canine seizures: similarities to human seizures. Epilepsy Behaviour 3, 460–470.
Licht, B.G., Lin, S., Luo, Y., Hyson, L.L., Licht, M.H., Harper, K.M., Sullivan, S.A., Fernandez, S.A. and Johnston, E.V. (2007) Clinical characteristics and mode of inheritance of familial focal seizures in Standard Poodles. Journal of the American Animal Hospital Association 231, 1520–1528.
Lohi, H., Young, E.J., Fitzmaurice, S.N., Rusbridge, C., Chan, E.M. et al. (2005) Expanded repeat in canine epilepsy. Science 307, 81.
Pákozdy, A., Leschnik, M., Sarchahi, A.A., Tichy, A.G. and Thalhammer, J.G. (2010) Clinical comparison of primary versus secondary epilepsy in 125 cats. Journal of Feline Medicine and Surgery 12, 910–916.
Pákozdy, A., Gruber, A., Kneissl, S., Leschnik, M., Halasz, P. and Thalhammer, J.G. (2011) Complex partial cluster seizures in cats with orofacial involvement. Journal of Feline Medicine and Surgery 13(10), 687–693.
Pákozdy, A., Halasz, P., Klang, A., Bauer, J., Leschnik, M., Tichy, A., Thalhammer, J.G., Lang, B. and Vincent, A. (2013) Suspected limbic encephalitis and seizure in cats associated with voltage-gated potassium channel (VGKC) complex antibody. Journal of Veterinary Internal Medicine 27(1), 212–214.
Panayiotopoulos, C.P. (2011) The new ILAE report on terminology and concepts for organization of epileptic seizures: a clinician’s critical view and contribution. Epilepsia 52, 2155–2160.
Panayiotopoulos, C.P. (2012) The new ILAE report on terminology and concepts for the organization of epilepsies: Critical review and contribution. Epilepsia 53(3), 399–404.
Patterson, E.E., Mickelson, J.R., Da, Y., Roberts, M.C., McVey, A.S., O’Brien, D.P., Johnson, G.S. and Armstrong, P.J. (2003) Clinical characteristics and inheritance of idiopathic epilepsy in Vizslas. Journal of Veterinary Internal Medicine 17, 319–325.
Penderis, J., Calvin, J., Abramson, C. et al. (2007) L-2-hydroxyglutaric aciduria: characterisation of the molecular defect in a spontaneous canine model. Journal of Medical Genetics 44, 334–340.
Podell, M. (2004) Seizures. In: Platt, S. and Olby, N. (eds) BSAVA Manual of Canine and Feline Neurology. BSAVA, pp. 97–112.
Podell, M., Fenner, W.R. and Powers, J.D. (1995) Seizure classification in dogs from a nonreferral-based population. Journal of the American Veterinary Medical Association 206, 1721–1728.
Poma, R., Ochi, A. and Cortez, M.A. (2010) Absence seizures with myoclonic features in a juvenile Chihuahua dog. Epileptic Disorders 12, 138–141. Epub 2010 May 20.
Quesnel, A.D., Parent, J.M., McDonell, W., Percy, D. and Lumsden, J.H. (1997) Diagnostic evaluation of cats with seizure disorders: 30 cases (1991-1993). Journal of the American Veterinary Medical Association 210, 65–71.
Rusbridge, C. (2005) Neurological diseases of the cavalier King Charles spaniel. Journal of Small Animal Practice 46, 265–272.
Schoeman, T., Williams, J. and Wilpe, E. (2002) Polyglucosan storage disease in a dog resembling Lafora’s disease. Journal of Veterinary Internal Medicine 16, 201–207.
Schriefl, S., Steinberg, T.A., Matiasek, K., Ossig, A., Fenske, N. and Fischer, A. (2008) Etiologic classification of seizures, signalment, clinical signs, and outcome in cats with seizure disorders: 91 cases (2000-2004). Journal of the American Veterinary Medical Association 233, 1591–1597.
Schwartz-Porsche, D. (1994) Seizures. In: Braund, K.G. (ed.) Clinical Syndromes in Veterinary Neurology, 2nd edn. Mosby, Missouri, pp. 238–251.
Schwartz-Porsche, D. and Kaiser, E. (1989) Feline epilepsy. Problems in Veterinary Medicine 1, 628–649.
Seppala, E.H., Jokinen, T.S., Fukata, M., Fukata, Y., Webster, M.T. et al. (2011) LGI2 Truncation Causes a Remitting Focal Epilepsy in Dogs. PLoS Genetics 7, e1002194.
Smith, M.O., Turrel, J.M., Bailey, C.S. and Gain, G.R. (1989) Neurologic abnormalities as the predominant signs of neoplasia of the nasal cavity in dogs and cats: seven cases (1973–1986). Journal of American Veterinary Medical Association 195, 242–245.
Stonehewer, J., Mackin, A.J., Tasker, S., Simpson, J.W. and Mayhew, I.G. (2000) Idiopathic phenobarbital-responsive hypersialosis in the dog: an unusual form of limbic epilepsy? Journal of Small Animal Practice 41, 416–421.
Thomas, W.B. (2010) Idiopathic epilepsy in dogs and cats. Veterinary Clinics of North America - Small Animal Practice 40, 161–179.
Webb, A.A., McMillan, C., Cullen, C.L., Boston, S.E., Turnbull, J. and Minassian, B.A. (2009) Lafora disease as a cause of visually exacerbated myoclonic attacks in a dog. Canadian Veterinary Journal 50, 963–967.
4 Reactive Seizures
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Reactive seizures are the reaction of a normal brain to a systemic metabolic, nutritional or exogenous toxic disorder (Podell, 1995). Reported prevalence of reactive seizures in dogs and cats varies among studies ranging from 7 to 32% in dogs and from 4 to 28% in cats (Bateman and Parent, 1999; Platt and Haag, 2002; Rusbridge, 2005; Pákozdy et al., 2008, 2010; Zimmermann et al., 2009; Brauer et al., 2011; Steinmetz et al., 2013) (see Chapters 7 and 8). In a recent study the most frequent cause of reactive seizures were intoxications (37/96, 39% of dogs) and hypoglycaemia (31/96, 32% of dogs) (see Fig. 7.2, Chapter 7); 49% (47/96) of dogs had generalized tonicclonic seizures with loss of consciousness and 41% (39/96) of dogs were presented in status epilepticus (Brauer et al., 2011). Dogs with reactive seizures caused by exogenous toxicity have a significantly higher risk to develop status epilepticus, particularly as first manifestation of a seizure disorder, than dogs with other seizure aetiologies (Zimmermann et al., 2009). In cats with reactive seizures and structural epilepsy, status epilepticus is significantly more common than in cats with idiopathic epilepsy (see Schriefl et al., 2008; Pákozdy et al., 2010; Chapter 3).
Clinical presentation in animals with systemic metabolic, nutritional and toxic disorders is variable depending on the underlying aetiology. Toxic disorders generally have an acute (less than 24 h) onset and neurological signs may be preceded or accompanied by gastrointestinal, cardiovascular or respiratory signs. However, chronic lead intoxication may result in recurrent seizures. Metabolic and nutritional disorders can present with an acute, subacute, or chronic onset and may be progressive or relapsing remitting. The neurological examination generally reveals diffuse, bilateral and often symmetrical neurological deficits, however seizures can sometimes be the only neurological abnormality. Diagnostic investigations (see Chapter 10) are aimed at identifying the underlying aetiology of the seizures and, if present, of the other clinical signs. Treatment is aimed at the underlying aetiology and seizure control with anti-epileptic medications (AEMs).
This chapter describes disorders commonly resulting in reactive seizures. For detailed information on specific AEMs, as well as management of cluster seizures and status epilepticus, the reader is referred to Chapters 12 to 24.
Systemic Metabolic Disorders Causing Seizures
Metabolic diseases can cause seizures by interfering with energy metabolism, altering osmolality, acid-base status, or producing endogenous toxins (O’Brien, 1998). The metabolic disorders most commonly reported to cause seizures in the veterinary literature are described below.
Hypoglycaemia
Overview
The blood glucose concentration is of prime importance for normal neuronal metabolism. Glucose is the single most important source of energy in the brain and carbohydrate storage in neural tissue is limited (Hess, 2010). Therefore cerebral function depends on a continuous supply of glucose. Glucose enters the brain by noninsulin dependent facilitated transport mechanisms, which requires a minimum blood glucose level to operate effectively. Persistent hypoglycaemia results in neuronal adenosine triphosphate (ATP) depletion, Na-K-ATPase dysfunction, cytotoxic oedema, excitatory neurotransmitter release (especially glutamate), activation of glutamate receptors, increased intracellular calcium, zinc, and nitric oxide synthase activity, reactive oxygen species generation, lipid peroxidation, DNA damage and cellular necrosis.
Clinical presentation
Hypoglycaemia at glucose concentrations of less than 60 mg/dl (<3 mmol/l) can result in neurologic signs including seizures. Clinical signs vary depending on the rate of decrease, magnitude and duration of hypoglycaemia and include behavioural changes, altered mental status, nervousness, tremors, generalized weakness and seizures. Severe brain damage, coma and death may occur with glucose concentrations of less than 18 mg/dl (<1 mmol/l). As with other homeostatically regulated parameters, the onset and severity of neurological signs may depend more on the rate of decrease than the actual concentration of glucose. Clinical signs may be triggered by fasting or exercise. There are multiple causes of hypoglycaemia (Box 4.1).
Diagnosis
The diagnosis of hypoglycaemic encephalopathy is based on documenting hypoglycaemia
Box 4.1. Underlying causes of hypoglycaemia.
Overproduction of endogenous insulin or insulin-like substances (e.g. insulin-like growth factor):
smooth-muscle tumours, hepatic tumours, or lymphoma.
Exogenous insulin overdose in animals with
diabetes mellitus (especially in cats).
Decreased glucose production:
toy or miniature breeds) and kittens.
Excess glucose consumption:
strenuous exercise.
Drug associated:
• Xylitol toxicity
in an animal with neurological signs that improve or resolve following normalization of blood glucose concentration. Specific diagnosis of the underlying disorder requires additional investigations. The reader is referred to internal medicine textbooks for further details on diagnosis and treatment of underlying aetiologies of hypoglycaemia. When hypoglycaemia is suspected in animals presenting with seizures or other neurological signs a blood sample should be taken. Falsely low glucose concentrations may result from use of human point-of-care glucometers in haemo-concentrated animals or from delayed separation of serum from blood cells (which continue to consume glucose).
Management
Once hypoglycaemia is confirmed, emergency treatment of hypoglycaemia-induced seizures involves slow (over 15 min) intravenous administration of 0.5–1 ml/kg of 50% dextrose, diluted 1:2 or 1:4 with 0.9% saline or sterile water. This dosage may need to be repeated and a continuous infusion of 5% dextrose (e.g. 50 ml of 50% dextrose in 500 ml of 0.9% saline or sterile water) may be necessary to maintain normoglycaemia. If intravenous therapy is difficult to perform, oral administration of a rapidly absorbed source of sugar (such as honey, Karo corn syrup or maple syrup) can be a useful substitute. Feeding frequent small meals can also help maintain normoglycaemia. Supportive care of the seizuring animal should be performed as described in Chapter 24. Specific treatment for the underlying cause of hypoglycaemia should be provided.
Insulinoma
Overview
Insulin secreting pancreatic b-islet cell neoplasia is the most common cause of hypoglycaemiainduced seizures in adult dogs. Feline insulinoma is rare. These tumours are associated with excess insulin secretion independent of the negative feedback effects caused by hypoglycaemia. Hyperinsulinaemia produces hypoglycaemia through suppression of glucose release and production rather than increased glucose utilization (Goutal et al., 2012). Most canine insulinomas are malignant. Metastatic lesions have been detected in 45% to 64% of dogs in different studies and most dogs have involvement of regional lymph nodes with or without distant metastases (commonly in the liver) by the time insulinoma is diagnosed (Hess, 2010). Insulinoma has been reported predominantly in adult to old dogs (mean age of 9 years) and in medium- to large-breed dogs.
Clinical presentation
Clinical signs are related to insulin-induced neuroglycopenia (seizures, generalized weakness, collapse, ataxia, obtundation and impaired vision) and/or to hypoglycaemia-induced catecholamine release (tremors, nervousness and hunger) (Goutal et al., 2012). Clinical signs can occur intermittently (Hess, 2010).
Fasting, excitement or exercise can precipitate or worsen clinical signs by decreasing blood glucose concentration. Feeding can result in either alleviation (when restoring normoglycaemia) or exacerbation (by stimulating insulin secretion) of clinical signs (Hess, 2010). A peripheral polyneuropathy has been described in dogs with insulinoma and may be caused by an immune mediated paraneoplastic process or by glucose-mediated metabolic disturbances in the lower motor neurons.
Diagnosis
Neurological signs consistent with hypoglycaemia (blood glucose concentrations below 60 mg/dl or 3 mmol/l) and concurrent hyperinsulinaemia (serum insulin concentration greater than 20 mU/ml) are suggestive of insulinoma. When a dog suspected of having an insulinoma is normoglycaemic, it should be fasted under close observation and blood glucose concentration should be measured every 1 to 2 h. Most dogs with insulinoma would develop hypoglycaemia within 12 h of fasting. When hypoglycaemia (blood glucose concentration <60 mg/dl or <3mmol/l) is detected, blood should be collected for measurement of insulin concentration and the dog should then be fed (Hess, 2010). Some dogs require repeated insulin measurements to confirm the suspicion of insulinoma. In one study hyperinsulinaemia was detected in 76% of dogs with insulinoma when insulin was measured once and in 91% of dogs when insulin was measured twice (Leifer et al., 1986). Fructosamine concentration can help to detect chronic hypoglycaemia as this parameter reflects the blood glucose concentrations over the previous 1–2 weeks and it has been reported to be significantly decreased in dogs with insulinomas. The clinical diagnosis of insulinoma can be further supported by identification of a pancreatic mass with abdominal ultrasonography or contrast-enhanced computed tomography (Figs 4.1, 4.2a, b). The sensitivity of abdominal ultrasonography in detecting insulinoma ranges from 28% to 75% (Goutal et al., 2012). Contrast-enhanced ultrasonography may increase the diagnostic yield of ultrasound. Ultrasound-guided fine-needle aspirate cytology represents a relatively noninvasive tool to


Fig. 4.2. Pre- and post-contrast CT images of the abdomen of a dog with an insulinoma in the left limb of the pancreas. On the pre-contrast image (a) the lateral aspect of the left limb of the pancreas is focally enlarged by a 15 mm diameter nodule (arrow). The nodule is slightly hypo-attenuating to adjacent normal pancreas. The post-contrast image (b) was obtained during the arterial phase of angiography. There is marked enhancement of the periphery and medioventral aspect of the mass. The dorsolateral aspect of the mass does not enhance (arrow). (Photo courtesy of Fraser McConnell, University of Liverpool)
Management
support the diagnosis of insulinoma. The definitive diagnosis is made following histo-Emergency treatment of hypoglycaemialogical examination of the tumour after sur-induced seizures involves slow intravenous gical resection. administration of 0.5–1 ml/kg of 50% dextrose, diluted 1:2 or 1:4 with sterile water, followed by an intravenous continuous rate infusion of 2.5–5% dextrose. Dextrose administration can be discontinued when clinical signs resolve.
Resolution of clinical signs may be difficult to achieve or to maintain in some dogs as the dextrose bolus may induce further insulin release from the tumour, leading to worsening of the hypoglycaemia. In such cases, repetitive dextrose boluses are not effective as a single strategy and either alternative or adjunctive therapies should be considered. These include: dexamethasone 0.1–0.5 mg/kg intravenously every 12 h, and glucagon at initial infusion rate of 5 ng/kg/min and subsequently adjusted based on blood glucose values up to 13 ng/kg/min. AEMs (see Chapters 12 and 24) may be necessary in severely affected cases. Frequent feeding of small meals should be initiated as soon as the animal can eat. Long-term treatment involves medical management (pre-and post-operatively or in dogs in which surgery cannot be performed) and surgical resection of the pancreatic mass and gross metastases. Whenever possible, surgical resection is considered the treatment of choice as despite being rarely curative, it offers the greatest chance of both durable control of clinical signs and prolonged survival time in dogs with insulinomas. However, outcome is affected by tumour staging. Some dogs can develop diabetes mellitus (transient or permanent) post-operatively. Medical management of insulinoma involves small frequent meals (every 4–6 h) of a diet rich in proteins, fat and complex carbohydrates, prednisolone
0.5 mg/kg/day orally (up to 4 mg/kg/day in refractory cases), and exercise restriction and avoidance of excitement. Additional medical therapy to relieve the hypoglycaemia may be required in some dogs and involves diazoxide 5 mg/kg every 12 h orally, which can be increased gradually without exceeding 60 mg/kg/day, or synthetic somatostatin such as octreotide (Goutal et al., 2012). Streptozotocin, a nitrosurea chemotherapeutic agent, can be used to selectively destroy beta cells in the pancreas or metastatic sites, but it can be nephrotoxic and emetogenic and its use in dogs warrants further investigations. At home immediate management of hypoglycaemic crisis involves oral administration of honey, corn syrup or maple syrup. Median survival time 12 to 14 months (range 0 days to 5 years) in dogs undergoing surgery in different studies. Whereas median survival ranges from 74 to 196 days in dogs undergoing medical treatment only. Combination of medical and surgical treatment resulted in a median survival time of 1316 days (44 months) in a recent study (Goutal et al., 2012).
Hepatic encephalopathy
Overview
Hepatic encephalopathy (HE) is a biochemical disorder of the brain secondary to various hepatic disorders such as congenital and acquired portosystemic shunt, micro-vascular dysplasia, congenital urea-cycle enzyme deficiencies, and acute or chronic severe parenchymal liver damage associated with cirrhosis, neoplasia, chronic active hepatitis, liver steatosis (lipidosis) in cats or toxicosis (Hardy, 1990). HE is more common in dogs than in cats.
Congenital portosystemic shunts represent the most common cause of HE. They generally occur as single vessels that provide direct vascular communication between the portal venous supply and the systemic venous circulation (caudal vena cava or azygous vein), bypassing the liver, and are not associated with portal hypertension. Approximately 25% to 33% of congenital portosystemic shunts are intrahepatic and the remainder are extrahepatic in dogs and cats (Berent and Tobias, 2009). The majority of intrahepatic portosystemic shunts occur in large or giant breed dogs, whereas most extrahepatic portosystemic shunts are seen in small and toy breed dogs (Berent and Tobias, 2009). In animals with congenital portosystemic shunts, the liver has been deprived of growth factors from birth and is therefore abnormally small with a hypofunctional parenchymal mass.
Acquired portosystemic shunts are usually multiple, extrahepatic and occur in animals with chronic portal hypertension secondary to hepatic arteriovenous malformations, noncirrhotic portal hypertension, or chronic hepatitis and cirrhosis (Berent and Tobias, 2009). Portosystemic shunts (congenital or acquired) occur far more commonly in dogs than in cats.
Hepatic microvascular dysplasia is a microscopic malformation of the hepatic microvasculature resulting in shunting of portal blood into the systemic circulation. Micro-vascular dysplasia can occur as an isolated disease or in association with macroscopic portosystemic shunts and can occur with or without concurrent portal hypertension. Hepatic microvascular dysplasia has been reported most commonly in the cairn terrier and Yorkshire terrier (Christiansen et al., 1995).
Urea-cycle enzyme deficiencies are rare congenital errors of metabolism in which one of the enzymes involved in ammonia metabolism fails.
Parenchymal hepatic diseases severely decrease the capacity of the liver to remove toxic products of intestinal metabolism and synthesize factors necessary for normal cerebral function.
The pathophysiology of HE is complex, multifactorial and incompletely understood (Box 4.2). The reader is referred to the cited references for a more detailed description of HE pathophysiology.
Clinical presentation
Neurological signs of HE are mainly consistent with forebrain involvement and include alterations in behaviour or personality, obtundation that may progress to stupor and coma, continuous pacing, aimless wandering, circling, head pressing, hypersalivation (particularly in cats), blindness and seizures. The neurological signs are often intermittent. Several factors can precipitate or exacerbate neurologic signs of HE (Box 4.3).
Non-neurological signs of HE vary depending on the underlying hepatic disease and include polyuria-polydipsia, vomiting, diarrhoea, weight loss, decreased endurance, ascites, icterus (rarely) and, in case of congenital portosystemic shunt, retarded or insufficient growth and signs of lower urinary tract disease (stranguria, pollakiuria, hematuria, dysuria) due to ammonium biurate crystalluria. Copper-coloured irises unusual for the breed have been reported in cats with congenital portosystemic shunts (Plate 1).
Diagnosis
Haematological changes include mild to moderate microcytic, normochromic nonregenerative anaemia. Serum biochemistry abnormalities
Box 4.2. Theories on hepatic encephalopathy pathophysiology (Maddison, 1992; Rothuizen, 2009; Bismuth et al., 2011; Poh and Chang, 2012; Kilpatrick et al., 2014).
which results in increased cholesterol uptake and synthesis of neurosteroids (e.g. allopregnanolone
and tetrahydradeoxycorticosterone) which have potent positive allosteric modulator properties on the GABAA receptor system;
L. De Risio
Box 4.3. Factors that can precipitate or exacerbate neurologic signs of HE.
hepatic metabolism.
include decreased albumin, urea, glucose and cholesterol levels. In animals with parenchymal hepatic disease, serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP) and total bilirubin are often elevated and vitamin K-dependent clotting factor levels may be decreased. Ammonium biurate crystals can be identified in the urine sediment in approximately half of the dogs with HE (Rothuizen, 2009).
The diagnosis of hepatobiliary disease can be confirmed by the presence of increased levels of fasting plasma ammonia and fasting
(e.g. 12-h) and post-prandial (e.g. 2-h) total serum bile acid concentrations. The sensitivity and specificity of fasting ammonia in the diagnosis of portosystemic shunts has been reported as 91% and 84% in dogs and 83% and 86% in cats, respectively (Ruland et al., 2010). The sensitivity and specificity of serum bile acids in the diagnosis of portosystemic shunts has been reported as 78% and 87% in dogs and 100% and 84% in cats, respectively (Ruland et al., 2010). Dogs and cats with congenital urea-cycle enzyme deficiencies have hyperammonaemia and low bile acid concentrations (Rothuizen, 2009).
Definitive diagnosis of portal vascular anomalies requires ultrasonography (Figs 4.3a–c), portovenography, scintigraphy (transcolonic or trans-splenic), computed tomographic angiography or magnetic resonance angiography (d’Anjou et al., 2004; Sura et al., 2007; Berent and Tobias, 2009; Mai and Weisse, 2011; Nelson and Nelson, 2011). Liver biopsy is required for confirmation of parenchymal hepatic disease. MRI of the brain of dogs and cats with portosystemic shunts may reveal cerebral cortical atrophy characterized by widened sulci and hyperintensity of the lentiform nuclei on T1-weighted images (Torisu et al., 2005). In addition, bilateral diffuse hyperintensity predominately of the parietal and occipital cortex grey matter on T2-weighted MR images has been reported in a dog with portosystemic shunt. The authors suggested that the signal changes represented cerebral oedema associated with cortical laminar necrosis caused by acute hyperammonaemia (Moon et al., 2012).
Management
The management of HE depends on the underlying cause, the degree of hepatic dysfunction and the severity of clinical signs. The aims of HE treatment are: (i) stop seizure activity; (ii) reduce serum levels of neurotoxic agents; and (iii) treat the underlying aetiology.
Seizures should be controlled with AEMs that undergo minimal or no hepatic metabolism such as levetiracetam (LEV) (see Chapter 16). LEV can be administered in dogs and cats at 60 mg/kg intravenously or orally as an initial loading dose, which can be followed (8h later) by a maintenance dose of 20 mg/kg every 8 h. Alternatively or in addition to LEV, bromide can be administered at a loading dose in dogs (see Chapter 14). Intravenous propofol as boluses (1–2 mg/kg) or constant rate infusion (0.1–0.6 mg/kg/min titrated to effect or up to 6 mg/kg/h) may be required in animals with persistent seizure activity (see Chapter 24). Oro-tracheal intubation, ventilation, haemodynamic support and intensive care monitoring may be required.
The production of ammonia in the gastrointestinal tract can be reduced by administration of:
• Lactulose (1–3 ml/kg/day orally divided into two to three doses) to decrease intestinal transit time, prevent constipation, alter the intestinal flora, and create an acid environment in order to trap ammonia as ammonium;
Reactive Seizures
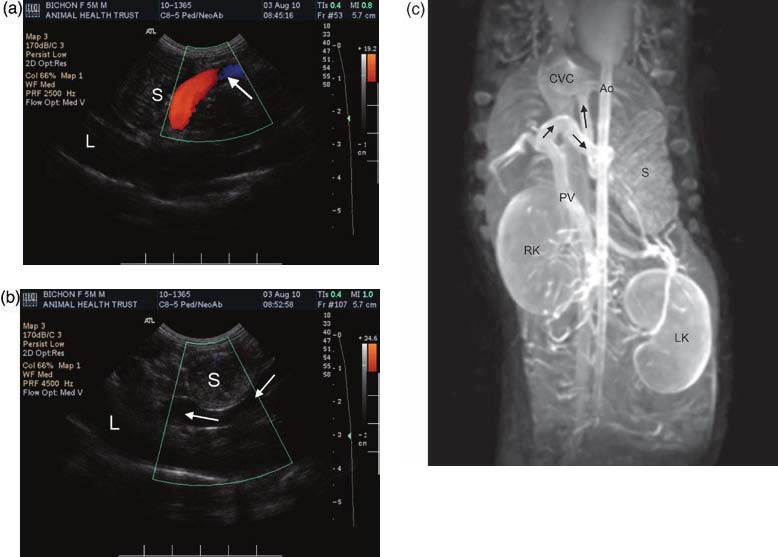
Fig. 4.3. Doppler ultrasound images (a, b) of the cranial dorsal abdomen of a 5-month-old male bichon frise with a portocaval shunt resulting in hepatic encephalopathy. Two images (a, b) are shown of different segments of the shunt at the level of the lesser curvature of the stomach (S). The shunt (arrows) is a large tortuous vessel arising from the right gastric vein. The liver (L) is small. MR angiography of the same dog (c). Blood is shunted via a large sigmoidal, tortuous vessel arising at the level of the gastroduodenal vein. The vessel loops (arrows) cranially then across the midline dorsal to the lesser curvature of the stomach and medial to the body of the stomach and finally loops cranially again to enter the caudal vena cava just caudal to the diaphragm close to the confluence of the left hepatic veins. RK, right kidney; LK, left kidney; PV, portal vein; S, stomach; Ao, aorta; CVC, caudal vena cava. (Photo courtesy of Andrew Halloway)
• Antibiotics such as ampicillin (22 mg/kg orally every 8 h), metronidazole (7.5 mg/ kg orally every 12 h), or neomycin (20 mg/ kg orally every 8 h), to reduce the colonic bacterial flora producing ammonia;
• Low-level high-quality protein diet.
In animals unable to take oral medications (e.g. propofol infusion, severe post-ictal phase, stupor or coma) lactulose can be administered by retention enema (20 ml/kg, made of three parts of lactulose diluted in seven parts warm water, every 4–6 h) and the antibiotic can be administered parenterally. Feline liver steatosis (lipidosis) should not be treated by dietary protein reduction but instead by forced feeding of amino acid-rich nutrients (Rothuizen, 2009). Protein intake should not be restricted excessively in growing young animals. A balanced rather than excessively restricted protein intake is currently preferred in people with HE (Bismuth et al., 2011). Administration of high doses of probiotics containing urease negative bacteria (such as Lactobacillus acidophilus or Enterococcus faecium) may help to modify the intestinal flora and decrease ammonia production. In addition, it has been suggested that administration of L-carnitine may reduce ammonia level by increasing energy metabolism (Bismuth et al., 2011). Intravenous fluids should be administered to animals with fluid and electrolyte abnormalities. Conditions that aggravate HE (see Box 4.3) should be avoided. Proton pump inhibitors such as omeprazole should be administered to animals with proximal gastrointestinal haemorrhage. Supportive nutraceutical therapy such as S-adenosyolL-methionine (SAMe) has been recommended for a variety of liver diseases; however, it is usually unnecessary in portovascular anomalies that can be treated surgically (Berent and Tobias, 2009). Congenital portosystemic shunts can be completely or partially ligated with nonabsorbable sutures or gradually attenuated with an ameroid constrictor, cellophane band, or hydraulic occluder. Gradual attenuation is preferred to reduce the risk of post- operative complications (Berent and Tobias, 2009). Acute complications of shunt attenuation include intraoperative haemorrhage, prolonged anaesthetic recovery, seizures and other forebrain neurological signs, portal hypertension and refractory hypoglycaemia. Severe persistent neurologic signs (including blindness and seizures) represent the most common cause of death after portosystemic shunts attenuation (Berent and Tobias, 2009). Central blindness has been reported in up to 44% of cats following portosystemic shunt surgery and usually resolves within 2 months after surgery (Kyles et al., 2002). Post-operative seizures have been reported in 3% to 7% of dogs and 8% to 22% of cats after shunt attenuation (Tisdall et al., 2000; Kyles et al., 2002; Frankel et al., 2006; Lee et al., 2006; Lipscomb et al., 2007). Seizures may occur up to 80 h after surgery, frequently progress to status epilepticus and may be difficult to control. These seizures are not associated with hypoglycaemia, hyperammonaemia, or attenuation technique (Berent and Tobias, 2009). Post-operative seizures can be treated with intravenous levetiracetam or propofol (see Chapters 16 and 24). Levetiracetam administered at 20 mg/kg orally every 8 h for a minimum of 24 h before surgery significantly decreased the risk of post-operative seizures and death in dogs undergoing surgical attenuation of extrahepatic congenital portosystemic shunts with ameroid ring constrictors (Fryer et al., 2011).
Renal-associated encephalopathy
Overview
Renal-associated encephalopathy can occur in animals with acute or end-stage chronic renal failure (uraemic encephalopathy), following haemodialysis (dialysis disequilibrium syndrome) or renal transplantation (post-renal transplantation encephalopathy). As for hepatic encephalopathy the pathogenesis of renal-associated encephalopathy is complex and incompletely understood. Neurological signs may result from accumulation of uraemic toxins, altered balance between excitatory and inhibitory neurotransmitters, as well as acid-base, fluid and electrolyte abnormalities. Parathyroid hormone (PTH) disturbance may also play a role in the pathogenesis of uraemic encephalopathy. In addition, hypertension secondary to renal disease may lead to intracranial cerebrovascular accidents (O’Brien, 1998). Dialysis disequilibrium syndrome is thought to be caused by an osmotic gradient between the brain cells and the extracellular fluid due to excessively rapid haemodialysis, which results in cellular swelling and cerebral oedema. Post-renal transplantation encephalopathy (resulting in seizures, stupor, ataxia and central blindness) has been reported in cats and it is most likely associated with uncontrolled hypertension (Kyles et al., 1999). Uraemic encephalopathy has been infrequently reported in dogs with renal failure (Wolf, 1980; Fenner, 1995).
Clinical presentation
Neurological signs of uraemic encephalopathy include obtunded mental status that can progress to stupor, muscle fasciculations (especially in facial muscles), head bobbing, generalized seizures and generalized weakness. In one retrospective study including 29 dogs, altered mental status was more common in dogs with chronic renal disease, whereas seizures were more common in dogs with acute renal failure (Fenner, 1995). Other clinical signs such as polyuria, polydipsia, vomiting and dehydration are reflective of renal failure.
Diagnosis
Diagnosis of renal-associated encephalopathy is based on signs of cerebral (mainly forebrain) dysfunction in an animal with renal failure or soon after haemodialysis or renal transplantation and no other cause of brain disease. Serum biochemistry shows very severe azotaemia, hyperkalaemia, and hyperphosphataemia with or without hypocalcaemia. Urinalysis performed before fluid therapy shows isosthenuria.
Management
Treatment should be aimed at the causes of the renal failure, if possible, as well as symptomatic and supportive. Fluid, electrolyte and acid-base imbalances should be corrected. Hypertension, if present, should be treated. Seizures can be treated as described in Chapters 12 and 24, however dosages of AEMs may need to be decreased in case of impaired renal excretion.
Hyponatraemia
Overview
Hyponatraemia occurs when plasma sodium concentration is below reference levels (usually 140 mEq/l or mmol/l in dogs, and 149 mEq/l or mmol/l in cats) (Benitah, 2010). Occurrence of clinical signs depends more on rapidity of onset of hyponatraemia than to magnitude of change. Neurologic signs may occur with sodium concentrations less than 120 mEq/l or mmol/l in dogs and less than 130 mEq/l or mmol/l in cats (de Morais and DiBartola, 2008a). Serum sodium concentration reflects the amount of sodium relative to the volume of water in the body and not total body sodium content. Hyponatraemic animals may have decreased, increased or normal total body sodium content and therefore volemic status also needs to be considered when investigating serum hyponatraemia (de Morais and DiBartola, 2008a). Hyponatraemia may result primarily from increased water gain, administration of fluids low in sodium (e.g. 5% dextrose in water, 0.45% sodium chloride), hypertonic solution without sodium
(e.g. mannitol, glucose) and an excessive sodium loss (e.g. loop diuretics, thiazides). Causes of hyponatraemia classified based on plasma osmolality and hydration status are listed in Box 4.4.
Clinical presentation
Clinical signs of hyponatraemia include lethargy, anorexia, vomiting, generalized weakness, ataxia, obtunded mental status progressing to stupor and coma, and seizures. The severity of neurologic signs is greater
Box 4.4. Causes of hyponatraemia classified
based on plasma osmolality and hydration status (modified from de Morais and DiBartola,
2008a).
With plasma hyperosmolality (>310 mOsm/kg):
nonsteroidal anti-inflammatory drugs,
vincristine);
• Myxedaema coma due to severe hypothyroidism;
• Hypovolaemia:
uroabdomen, cavitary effusion);
administration).
when hyponatraemia develops rapidly. In acute hyponatraemia, water flows down its concentration gradient and enters brain cells producing cerebral oedema and increased intracranial pressure. In addition, hypervolemic animals may be presented with ascites, peripheral or pulmonary oedema, and jugular distension; whereas hypovolemic animals may present with signs of dehydration including decreased skin turgor, dry mucous membranes, delayed capillary refill time, tachycardia, hypotension, increased packed cell volume (PCV) and total protein concentration, and high urine specific gravity. Determination of the animal’s volume status helps to identify the underlying cause of the hyponatraemia and initiate correct treatment.
Diagnostic investigation
In hypovolemic and hyponatraemic animals calculation of the fractional excretion of sodium (FENa) can be used to determine if the kidneys are the source of excessive sodium loss (see equation below). The FENa should be less than 1% for nonrenal sources of sodium loss and 1% or greater if sodium is being lost by the kidneys (de Morais and DiBartola, 2008a).
Calculation of the fractional excretion of sodium is:
U /S
Na Na
FENa = ´ 100 (4.1)
U /S
Cr Cr
where FENa = fractional excretion of sodium, UNa = urine concentration of sodium (mEq/l), SNa = serum concentration of sodium (mEq/l), UCr = urine concentration of creatinine (mg/dl) and SCr = serum concentration of creatinine (mg/dl).
Additional diagnostic investigations including laboratory analysis and imaging will vary depending on the suspected underlying aetiologies of hyponatraemia (see Box 4.4). The reader is referred to internal medicine textbooks for further details on diagnosis and treatment of each condition.
Management
Treatment is aimed at increasing serum sodium levels and treating the underlying cause of the hyponatraemia. Isotonic (0.9%) saline or balanced electrolytes solutions can be administered to hypovolemic animals, while water restriction (i.e. limiting water intake to less than urine output) can be performed for animals with normovolaemia or hypervolaemia associated with excessive water intake or renal retention. A loop diuretic and dietary sodium restriction can be considered in hypervolaemic animals. To avoid life-threatening neurologic complications such as brain stem myelinolysis, recommended rates of correction for chronic (>2 days) hyponatraemia are 10–12 mEq/l/day or approximately 0.5 mEq/l/h (0.5 mmol/l/h) (Benitah, 2010). When hyponatraemia is corrected too rapidly, the increasing osmolality of the extracellular space results in water movement from the intracellular to the extra-cellular space, and consequent cellular dehydration and shrinkage. This cellular shrinkage can separate the neurons from their myelin sheaths leading to myelinolysis. Mental status, hydration and electrolytes (particularly serum sodium concentration) need to be monitored frequently (e.g. every 4–6 h) during correction of hyponatraemia. Seizure can be treated as described in Chapters 12 and 24, however dosage and choice of AEMs will vary depending on the underlying aetiology of hyponatraemia.
Hypernatraemia
Overview
Hypernatraemia occurs when plasma sodium concentration is above reference levels (usually 155 mEq/l or mmol/l in dogs, and 162 mEq/l or mmol/l in cats) (Benitah, 2010). Occurrence of clinical signs depends more on rapidity of onset of hypernatraemia than to magnitude of change. Neurological signs generally occur when serum sodium levels exceed 170 mEq/l or mmol/l in dogs and 175 mEq/l or mmol/l in cats (>350 mOsm/kg) (de Morais and DiBartola, 2008b). Hypernatraemia can result from water or hypotonic fluid loss, or excessive sodium gain (Box 4.5).
Sodium and its attendant anions account for approximately 95% of the osmotically active
Box 4.5. Causes of hypernatraemia (modified from de Morais and DiBartola, 2008b).
Pure water deficit (normovolaemic hyper
natraemia):
• Inadequate water intake;
• Diabetes insipidus (central or nephrogenic).
Hypotonic fluid loss (hypovolaemic
hypernatraemia):
• Osmotic diuresis (mannitol infusion,
hyperglycaemia);
• Burns.
Excessive sodium gain (hypervolaemic
hypernatraemia):
• Hypertonic fluid administration (intravenous
hypertonic saline, sodium bicarbonate, sodium
phosphate enema);
poisoning).
substances in the extracellular water. Therefore hypernatraemia is associated with hyperosmolality (de Morais and DiBartola, 2008b).
Clinical presentation
Clinical signs of hypernatraemia are mainly neurological and include anorexia, lethargy, vomiting, behavioural changes, head pressing, ataxia, generalized muscular weakness, muscle fasciculations, seizures, blindness, obtunded mental status progressing to stupor and coma, and death in severe cases. The severity of the neurological signs depends more on the rate of increase in sodium concentration than on the degree of hypernatraemia. With acute hypernatraemia water moves out of cells into the hyperosmolar extracellular space producing neuronal dehydration. The resulting decrease in cerebral volume may cause stretching and tearing of small cerebral vessels, leading to intracranial haemorrhage (subarachnoid, subdural, and/or intraparenchymal). Both cellular dehydration and intracranial haemorrhage may contribute to cerebral dysfunction in the acutely hypernatraemic animal (Benitah, 2010). When hypernatraemia develops slowly and gradually (e.g. <1 mEq/l/h or <1 mmol/l/h), the cerebral neurons compensate by increasing intracellular osmolality by movement of sodium, potassium, chloride and glucose intracellularly and by producing osmotically active solutes, called idiogenic osmoles (such as taurine, sorbitol and inositol) to adapt to the hypertonicity and minimize cerebral cellular dehydration. Clinical signs may be minimal or absent with slowly developing hypernatraemia. In addition to the clinical signs caused by the hypernatraemia and associated hyperosmolality, clinical signs of hypovolaemia (decreased skin turgor, dry mucous membranes, delayed capillary refill time, tachycardia, hypotension, increased PCV and total protein concentration, and high urine specific gravity) or hypervolaemia (ascites, peripheral or pulmonary oedema and jugular distension) may be present.
Diagnosis
Diagnostic investigations including laboratory analysis and imaging will vary depending on the suspected underlying aetiologies of hypernatraemia (see Box 4.5). The reader is referred to internal medicine textbooks for further details on diagnosis and treatment of each condition.
Management
Treatment is aimed at restoring normal extra-cellular fluid volume, decreasing sodium serum levels and treating the underlying cause of the hypernatraemia. Serum sodium concentration should be corrected at a rate of less than 0.5 mEq/l/h (0.5 mmol/l/h) to minimize the risk of cerebral cellular swelling, cerebral oedema and increased intracranial pressure. Free water deficit can be calculated based on the formula in equation 4.2 at bottom of page.
Oral water administration is the preferred method to correct water deficits in normovolaemic animals. Isotonic intravenous fluids should be used in normovolaemic animals that cannot drink and in hypovolaemic animals. Once the extracellular fluid volume has been restored, hypotonic fluids can be administered as maintenance treatment. Normovolaemic animals with pure water deficits can be administered 5% dextrose in water intravenously. Hypernatraemia secondary to excessive sodium gain can be treated with 5% dextrose in water intravenously and a loop diuretic to promote natriuresis (Benitah, 2010). Mental status, hydration and electrolytes (particularly serum sodium concentration) need to be monitored frequently (e.g. every 4–6 h) during correction of hypernatraemia. Seizures can be treated as described in Chapters 12 and 24; however, dosage and choice of AEMs will vary depending on the underlying aetiology of hypernatraemia.
Hypocalcaemia
Overview
Hypocalcaemia results in tetany (intermittent contraction of extensor muscles) and seizures when serum ionized calcium concentration is equal or lower than 0.8 mmol/l (3.2 mg/dl) or total calcium concentration is below 1.5 mmol/l (6 mg/dl) (Drobatz and Casey, 2000; Brauer et al., 2011). To convert mmol/l to mg/dl, the value in mmol/l has to be multiplied by 4 (Schenck and Chew, 2008). Total serum calcium is approximately 50% ionized, 40% protein bound (especially to albumin), and 10% chelated with anions such as citrate or phosphate. Only ionized calcium is biologically active. The proportion of ionized calcium is affected by serum protein level, acid-base status (ionized calcium levels are decreased by alkalosis and increased by acidosis) and the presence of anions that act as chelators (e.g. Mg++ or phosphate). Calcium homeostasis is regulated by parathyroid hormone (PTH), calcitriol (1,25- dihydroxyvitamin D) and calcitonin. The main organs involved in calcium metabolism are bone, kidney and small intestine.
Low calcium concentrations increase neuronal membrane permeability to sodium ions, resulting in neuronal hyperexcitability in the peripheral and central nervous system.
Clinical presentation
Clinical signs include nervousness, behavioural abnormalities (aggression, vocalization), decreased activity, panting, pacing, muscle stiffness, fasciculations, cramping and tetany, shifting limb lameness, stiff gait, hyperthermia, facial rubbing and biting at feet (probably due to paraesthesia) and seizures. Neurological signs can be intermittent and sometimes can be triggered by external stimuli or exercise. Differential diagnoses for underlying aetiologies of hypocalcaemia in dogs and cats are listed in Box 4.6. The reader is referred to internal medicine textbooks for further details on diagnosis and treatment of each condition.
Diagnosis
Diagnosis of hypocalcaemia is based on characteristic clinical signs and serum-ionized calcium concentration equal or lower than 0.8 mmol/l (3.2 mg/dl). Decreased serum total calcium should prompt assessment of serum ionized calcium. The use of corrective formulae to estimate ionized calcium concentration based on serum total calcium and total protein or albumin concentration is not recommended. These formulae do not accurately predict ionized calcium concentration and should not be used to make therapeutic decisions. Serum
Free water deficit (l) = 0.6 × lean body weight (kg) × ( (plasma sodium/140) −1) (4.2)
Box 4.6. Causes of hypocalcaemia (modified from Schenck and Chew, 2008).
ionized calcium concentration is typically higher than ionized calcium concentration in heparinized plasma or whole blood (measured by means of a blood gas analyser) due to dilution with heparin (Schenck and Chew, 2008). Calcium concentrations should not be assessed on EDTA plasma as EDTA chelates calcium, giving artificially low calcium concentrations. Additional laboratory investigations (such as measurement of plasma PTH, magnesium, vitamin D metabolites, urinalysis) and diagnostic imaging are indicated depending on the suspected underlying cause of hypocalcaemia in individual patients.
Management
Diazepam (0.5 to 1.0 mg/kg intravenously, up to a maximum total dose of 20 mg, in dogs and cats, repeated to effect or twice within 2 h; see Chapters 21 and 24) can be used to control the tetany and seizures initially while the diagnosis of hypocalcaemia is confirmed. Hypocalcaemia is treated with 10% calcium gluconate at a dose of 0.5–
1.5 ml/kg (providing 5–15 mg/kg of calcium) administered slowly IV (over 10 to 30 min) to effect as individual requirements vary. Clinical improvement is generally obvious within minutes of initiating the infusion. Electrocardiographic monitoring is advisable because of the risk of cardiotoxicity. If bradycardia, premature ventricular complexes, increased P-R interval, prolonged QRS complex or shortening of the Q-T interval is observed, the IV infusion should be briefly discontinued. Maintenance therapy of hypocalcaemia involves calcium gluconate (at a dosage equal to the one used IV for the control of tetany), diluted in an equal volume of saline, administered SC every 6 to 8 h. Serum calcium concentration should be monitored every 1–3 days to adjust the calcium gluconate dose. Generally, after serum calcium concentrations have been maintained within reference range for 48 h, the administration of calcium gluconate can be gradually tapered off to every 8–12 h. Long-term maintenance treatment with oral calcium (25 mg/kg every 8–12 h) and vitamin D supplementation may be required. Specific treatment for the underlying aetiology of hypocalcaemia should be instituted as promptly as possible.
Nutritional Disorders Causing Seizures
Thiamine (vitamin B1) deficiency
Overview
Thiamine (vitamin B1) deficiency has been reported in dogs and cats fed thiamine-deficient diets including commercial canned pet food (Marks et al., 2011; Markovich et al., 2014). Thiamine can be destroyed by heat during cooking or processing, preservatives such as sulfur dioxide, sulfate trace minerals, ultraviolet and gamma irradiation, and thiaminase enzyme activity, which is found predominantly in shellfish, fish viscera and some bacteria. In addition, thiamine deficiency can result from decreased intake due to anorexia or vomiting, decreased intestinal absorption (e.g. diarrhoea), abnormal utilization
(e.g. liver dysfunction) or increased requirements (e.g. fever, infection). The metabolically active form of thiamine, thiamine pyrophosphate, plays an essential role in three enzyme systems (pyruvate dehydrogenase, alpha Tissues dependent on glucose or lactateketoglutarate dehydrogenase, and transketo-pyruvate for energy, such as the brain and lase), which are essential for complete oxida-heart, are particularly compromised in thiation of glucose through the Krebs cycle. mine deficiency.
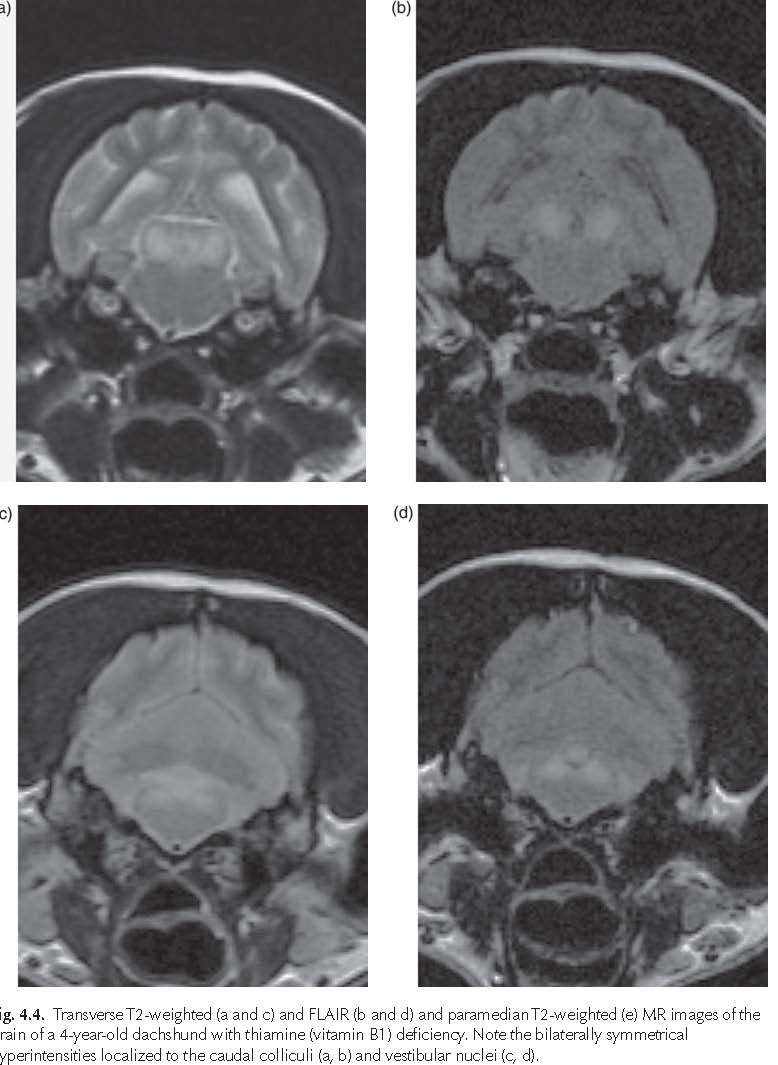


Fig. 4.5. Transverse T2-weighted MR image of the brain of a domestic short hair cat with thiamine (vitamin B1) deficiency. Note the bilaterally symmetrical hyperintensities localized to the lateral geniculate nuclei. See Plate 2 for the histology showing focal haemorrhagic necrotic lesions localized to the lateral geniculate nuclei.
L. De Risio
Clinical presentation
Clinical signs include anorexia, vomiting, abnormal mentation, seizures, dilated and unresponsive pupils, absent menace response bilaterally, opisthotonos with increased extensor tone of all four limbs, ataxia, tetraparesis, postural reaction deficits and vestibular dysfunction (Garosi et al., 2003; Penderis et al., 2007; Palus et al., 2010). In addition, cervical ventroflexion and hyperesthesia to stimuli have been reported in thiamine-deficient cats. Seizures can progress to coma and death if the thiamine deficiency is not treated.
Diagnosis
A presumptive diagnosis of thiamine (vitamin B1) deficiency is based on dietary history, clinical and MRI findings and response to therapy. MRI findings include bilaterally symmetrical hyperintensity on T2-weighted and FLAIR images localized to the red nuclei, caudal colliculi, vestibular nuclei and cerebellar nodulus in dogs (Figs 4.4a–e) (Garosi, 2003) and to the lateral geniculate nuclei, caudal colliculi, periaqueductal grey matter, medial vestibular nuclei, cerebellar nodulus and facial nuclei in cats (Fig. 4.5; Plate 2) (Penderis et al., 2007; Palus et al., 2010). These MRI changes have been reported to resolve following thiamine supplementation (Garosi et al., 2003; Palus et al., 2010). Bilaterally symmetrical hyperintense lesions on T2-weighted and FLAIR images have been reported to affect also the cerebral cortex (parietal, occipital, hippocampal lobe) in cats with thiamine deficiency (Marks et al., 2011). Diagnosis can be confirmed by determining whole blood thiamine concentration by high-performance liquid chromatography. This test is now commercially available in many countries and has replaced the erythrocyte transketolase activity assay because of its superior sensitivity and specificity for thiamine status (Marks et al., 2011). Diet samples can also be submitted for thiamine analysis. Pathologic changes include bilaterally symmetrical spongiosis, necrosis and haemorrhage of upper brainstem nuclei including caudal colliculus, lateral geniculate (Plate 2), medial vestibular and oculomotor nuclei.
Management
Treatment should be instituted immediately for any animal suspected of having thiamine deficiency. Thiamine should be administered at 50–100 mg per dog and 25–50 mg per cat IM, SC or PO every 12–24 h until a response is obtained or another diagnosis is established. Emergency management of seizures can be performed as described in Chapter 24. The underlying cause of thiamine deficiency should be identified and managed. The majority of affected dogs and cats respond rapidly to thiamine supplementation and diet change.
Exogenous Toxic Disorders Causing Seizures
Overview
Numerous exogenous toxins can induce seizures (see Box 3.1, Chapter 3) through different mechanisms including increased excitation, decreased inhibition, and interference with energy metabolism (O’Brien, 1998). Toxins may affect the nervous system directly or indirectly, by affecting other organs whose dysfunction secondarily affects the brain (e.g. HE due to toxin-induced hepatic failure, xylitol-induced hypoglycaemia). This chapter focuses predominantly on toxins with direct effect on the nervous system.
Clinical presentation
The suspicion of toxin exposure is often based on the history and the onset of acute neurological signs (including excitation and hyperactivity or obtundation, stupor, coma, muscle tremors and fasciculations, seizures and ataxia) often associated with vomiting, diarrhoea, salivation, bronchoconstriction, bradycardia or tachycardia and hyperthermia. The source of intoxication is not always obvious to the pet owner, and therefore veterinarians should be proactive in asking questions and mentioning possible sources of intoxication any time the clinical presentation raises the suspicion of toxin exposure. Seizure can occur in clusters or as status epilepticus (e.g. organophosphates,
Reactive Seizures
strychnine, mycotoxins) or less commonly may be isolated and recurrent (e.g. lead) (O’Brien, 1998).
Diagnosis
The presumptive diagnosis is often based on the history of toxin ingestion and clinical signs. Definitive diagnosis is made by identification of the toxin in feed or suspect bait material, gastric content from vomitus or lavage fluids, water, blood or urine (for urinary excreted toxins).
Management
Emergency treatment of neurotoxicity involves multiple simultaneous steps, including:
Decontamination
The methods of decontamination and prevention of further toxin absorption depend on the route of entry of the toxin and its metabolic profile.
Cutaneous absorbed toxins
Bathing is the standard method of decontamination for cutaneous exposure to most toxic substances (Rosendale, 2002). The patient should be stabilized prior to bathing. The stimulation of bathing may trigger or exacerbate neurological and cardiovascular signs. People handling the animal should wear protective gloves and aprons. Liquid handdishwashing detergents are usually recommended over shampoo as they are more effective at removing lipid soluble substances. Sometimes bathing must be repeated to completely remove the toxin. Clipping may be the best way to remove adhesive substances.
Gastrointestinal absorbed toxins
When the toxin has been ingested, decontamination involves:
Emetics that stimulate vomiting by direct irritation of the pharyngeal or gastric mucosa should not be used. Induction of emesis is contraindicated in case of ingestion of caustics or volatile substance, if vomiting has already occurred, and in animals with seizures, impaired mental status or abnormal gag reflex due to the risk of aspiration pneumonia (Rosendale, 2002).
Gastric lavage is performed in the anaesthetized animal by gastric intubation and irrigation of the stomach with warm water. A cuffed endotracheal tube must be in place during the procedure to minimize the risk of aspiration of stomach contents. Gastric lavage is indicated when induction of emesis is contraindicated (e.g. profound CNS depression or seizures) or when a large amount of toxin is likely to still be present in the stomach. Gastric lavage is contraindicated in the case of ingestion of: caustics, small volume of toxin (which can be decontaminated with activated charcoal) and moderate amount of toxin more than 2 h previously.
Table 4.1. Medications used in animals with exogenous toxic disorders.
Indication Medication Dosage
Seizure control Diazepam 0.5 to 1.0 mg/kg IV, up to a maximum total dose of 20 mg, or
0.5 to 2.0 mg/kg intrarectally in dogs and cats, repeated to effect or twice within 2 h; if seizures persist, diazepam can be administered as constant rate infusions of 2–5 mg/h in 5% dextrose in water (see Chapter 21)
Levetiracetam 20–60 mg/kg IV, IM, PO once, followed after 8 h by 20 mg/kg IV, IM, PO q8h (see Chapter 16)
Phenobarbital 15–20 mg/kg IV, IM or PO divided in multiple doses of 3–5 mg/kg over 24–48 h, and if seizures persist, followed by 2–3mg/kg every 12 h in dogs, and 1.5-2.5 mg/kg every 12 h in cats (see Chapter 13)
Propofol 1–4 mg/kg IV bolus or 0.1–0.6 mg/kg/min constant rate infusion titrated to effect or up to 6 mg/kg/h (see Chapter 24)
Skeletal muscle Methocarbamol Initial dose 44–220 mg/kg IV given in small boluses of relaxation 30–40 mg/kg to effect, up to 330 mg/kg in 24 h in dogs
(control of and cats. Oral administration is also an option
tremors) Diazepam 0.5 to 1.0 mg/kg IV to effect or PO, up to a maximum total dose of 20 mg in dogs and cats, up to three times in 24 h
Induction Apomorphine 0.04 mg/kg IV or 0.06 mg/kg SC or IM, in dogs of emesis Xylazine 0.4 mg/kg, IM or SC, in cats
Prevention Activated charcoal 1–5 g/kg of activated charcoal solution or of powdered of further activated charcoal mixed with 50 to 200 ml of water to absorption make a slurry
of toxin Sodium sulphate 250 mg/kg PO once to a maximum of 5 g in cats and 25 g (40% solution) in dogs Sorbitol 1 to 2 ml/kg (0.7–1.4 g/kg) PO once (70% solution)
Reduction of free 20% lipid 1.5 ml/kg IV slow (over 2–15 min) bolus followed by a and tissue levels preparation for constant rate infusion of 0.25 ml/kg/min for 30 to 60 min. of lipophilic intravenous infusion This may be repeated every 4 h as long as serum is not agents lipaemic but should be discontinued if a positive
response is not seen after three treatments. Animals should be hospitalized and monitored until clinical signs
have resolved and the serum is no longer lipaemic as
signs of toxicity may return after the intravenous lipid emulsion has been metabolized
The risks of general anaesthesia and of aspiration pneumonia need to be considered. The animal must be monitored continuously during recovery from general anaesthesia from gastric lavage.
Colonic lavage is indicated for toxins than may be absorbed from the colon when ingestion has occurred more than 4–6 h before presentation. Colonic lavage may be beneficial when performed earlier than 4 h post-ingestion of organophosphates and carbamates. Colonic lavage is performed by inserting a lubricated narrow non-rigid tube from the anus into the rectum and transverse colon and instillation of warm water under gravity flow. Animals with decreased consciousness should be endotracheally intubated during the procedure as colonic distension can stimulate emesis.
Activated charcoal can be administered by stomach tube after gastric lavage or syringe-fed to animals that can swallow. Activated charcoal is indicated for adsorption of most toxins when the toxic substance is likely to still be present in the gastrointestinal tract, particularly with toxins with slow gastrointestinal release and adsorption or toxins undergoing enterohepatic recirculation. In these cases repeated dosing of activated charcoal (without sorbitol) 1–4 g/kg PO every 6–8 h for 24 h is indicated. Serum sodium should be closely monitored for patients receiving repeated charcoal doses due to potential of development of hypernatraemia. Constipation can be prevented by maintaining the animal well hydrated. Strong acids or alkalis, dissociable salts and metals, and alcohols are not adsorbed by activated charcoal. Activated charcoal is contraindicated in animals with high risk of aspiration pneumonia or hypernatraemia. Cathartics are often administered in association with activated charcoal to minimize toxin absorption by reducing intestinal transit time. Repeated dosing is not recommended due to the risk of osmotic diarrhoea and hypernatraemia. Cathartics are contraindicated in dehydrated or hypovolaemic animals.
Urinary excreted toxins
With urinary excreted toxins, diuresis and/or modification of the urine pH can enhance toxin excretion (O’Brien, 1998). Urine acidification can be achieved by administering ammonium chloride, 100 mg/kg PO for dogs and 20 mg/ kg PO for cats. Ammonium chloride should not be used in acidotic animals and overuse may result in ammonia toxicosis. Urine alkalinization can be achieved by administering sodium bicarbonate at 0.5 to 2 mEq/kg IV every 4 h. Possible adverse effects of systemic pH modulation need to be considered. The acid-base status of the animal must be monitored.
Intravenous lipid emulsion infusion
Intravenous lipid emulsion (IVLE) (e.g. Intralipid) infusion has been increasingly used as antidotal treatment of toxicosis from various lipophilic agents (Fernandez et al., 2011; Gwaltney-Brant and Meadows, 2012; Kaplan and Whelan, 2012). IVLE is composed of neutral, medium to long-chain triglycerides derived from plant oils (e.g. soybean, safflower), egg phosphatides and glycerine, formulated primarily as a source of essential fatty acids for patients requiring parenteral nutrition (Gwaltney-Brant and Meadows, 2012). The exact mechanism of antidotal action of IVLE is unknown. The sequestration effect theory proposes that IVLE acts as pharmacological ‘sink’ for lipid soluble compounds decreasing their tissue availability and increasing their clearance (Kaplan and Whelan, 2012). IVLE expands the plasma lipid phase creating a discrete compartment that sequesters lipophilic agents and prevents them from reaching their sites of action. In addition, the expanded plasma lipid phase creates a concentration gradient that facilitates the passage of the lipophilic compounds from the interstitial space into the intravascular space. Potential adverse effects of IVLE infusions include: interference with lipophilic medications (i.e. methocarbamol, diazepam, phenobarbitone, propofol) administered for symptomatic or supportive care; pancreatitis due to persistent lipaemia; and hypersensitivity due to IVLE components (Gwaltney-Brant and Meadows, 2012). Based on growing number of case reports in veterinary medicine, IVLE infusion shows promise in the management of toxicosis from a variety of lipophilic agents, including macrocyclic lactones and pyrethrin compounds (Pritchard, 2010; Clarke et al., 2011; Haworth and Smart, 2012; Epstein and Hollingsworth, 2013). More studies are needed to determine optimum time of initiation, dosing regimens and margin of safety of IVLE as antidotal treatment for different lipid soluble toxicities. Treatment protocols are likely to be affected by the degree of lipid solubility and half-life of the toxin. The protocol recommended in Table 4.1 is based on the human literature and veterinary case reports. Care must be taken to use aseptic technique when handling and administering any lipid emulsions as they can promote bacterial growth (Kaplan and Whelan, 2012).
Insecticides
Pyrethrin and pyrethroid (permethrin)
Overview
Pyrethrins are natural insecticides obtained from Chrysanthemum cinerariaefolium, while pyrethroids (e.g. permethrin and fenvalerate) are synthetic analogues of pyrethrins classified as type I (no alpha-cyano-3-phenoxybenzyl group) or type II (with alpha-cyano-3phenoxybenzyl group) (Hansen, 2006; Wismer and Means, 2012). Etofenprox is a nonester pyrethroid-like insecticide. Permethrin toxicosis is one of the most commonly reported poisonings in the USA and the UK in small animals. Pyrethrins and pyrethroids can be absorbed dermally, orally (e.g. grooming in cats) and via inhalation. Many pyrethrin and pyrethroid formulations are registered for topical use on dogs and/or cats (as spot-on, flea collars, medicated shampoo) and in the household for flea and tick control. Generally, most products registered for use on dogs and cats are safe when used according to label directions in healthy pets. Cats are more sensitive than most other species to pyrethrins and pyrethroids probably due to deficiencies in glucoronyl transferase resulting in slower hepatic metabolism of these compounds. Intoxication results from administration to cats of products labelled for use on dogs, overdose or repeated over-application (Hansen, 2006). Secondary exposure may occur in cats that are in contact with dogs or treated environments. Pyrethrins and pyrethroids are highly lipophilic and rapidly distribute to adipose tissue and the central and peripheral nervous system (Wismer and Means, 2012).
Mechanism of action
Type I and II pyrethroid and pyrethrin compounds can slow both opening and closure of voltage-gated sodium channels, causing prolonged neuronal depolarization or repetitive discharges of motor and sensory nerve fibres. Type II pyrethroids also inhibit binding of GABA to the GABAA receptor, which prevents influx of chloride. This causes further membrane depolarization, blockade of action potential and failure of membrane repolarization.
Clinical presentation
Clinical signs of toxicosis may occur within 3 to 72 h of exposure and include muscle fasciculations and tremors, hypersalivation, ataxia, vomiting, diarrhoea, obtundation, mydriasis, hyperexcitability, hyperactivity, paresthesia, hyperesthesia, seizures and dyspnoea. Type I pyrethroids tend to cause tremors and seizures, whereas type II pyrethroids cause depolarizing conduction blocks with weakness and paralysis (Wismer and Means, 2012). Death may occur but is uncommon.
Diagnosis
Clinical diagnosis is based on history of exposure and clinical presentation. The pet-owners should be specifically questioned on recent ectoparasite treatment either directly on the affected animal or other pets in the house.
Management
Treatment is symptomatic and supportive, including dermal decontamination by bathing with a mild hand-dishwashing detergent and water (in case of dermal exposure to spot-on preparations), methocarbamol (Table 4.1), AEMs (see Table 4.1 and Chapters 12 and 24), fluid therapy and oxygenation (if necessary). Activated charcoal (Table 4.1) can be used in case of oral exposure. The use of intravenous lipid emulsion (Table 4.1) as adjunctive treatment to reduce tissue concentrations of permethrin has produced encouraging results in cats with permethrin toxicosis (Haworth and Smart, 2012; Kuo and Odunayo 2013).
Paresthesia to spot-on preparations may be treated by rubbing vitamin E, corn or olive oil on the application area (Wismer and Means, 2012).
Prognosis
Clinical signs generally resolve within 72 h following appropriate treatment (Hansen, 2006). However, recovery may take a week or longer in some cases. Delayed treatment and generalized seizures are associated with a less favourable prognosis and increased probability of death. Mortality rates vary between 5% and 45% in cats.
Organophosphates and carbamates
Overview
Organophosphates (chlorpyrifos, diazinon, dichlorvos, fenthion) and carbamates (aldicarb, methomyl, carbofuran, carbaryl) are widely used for insect and nematode control in dogs and cats and for insect control in the household and garden. They are available as sprays, pour-ons, oral anthelminthics, baits, collars, dips, dusts and granules (Wismer and Means, 2012). Exposure commonly results from accidental cutaneous overdose or ingestion (Dorman and Fikes, 1993). Organophosphate toxicity may result in acute (<7 h), intermediate (7–96 h) and delayed (1–4 weeks) syndromes.
Mechanism of action
Organophosphates are irreversible inhibitors of acetylcholine esterase (AChE) and recovery depends upon synthesis of new AChE. Carbamates are reversible inhibitors of AChE with restoration of AChE activity when the carbamate insecticide and enzyme separate. Organophosphate and carbamate intoxication results in accumulation of the neurotransmitter acetylcholine in the synaptic cleft and activation of muscarinic, nicotinic and CNS cholinergic synapses.
Clinical presentation
Clinical signs of acute organophosphate toxicity develop within minutes to hours depending on dose, route and toxicity of the compound and include:
All of the above clinical signs are not necessarily seen in every case and variability depends on toxicant type, dosage, formulation, route of exposure, poisoned species and stage of intoxication. Cats are generally more susceptible to AChE inhibitors than dogs. Death from either organophosphates or carbamates is associated with respiratory dysfunction resulting from respiratory tract secretions, bronchiolar constriction, intercostal and diaphragm muscle paralysis and CNS-mediated respiratory paralysis.
The intermediate and chronic (also named organophosphate-induced delayed neuropathy) syndromes are characterized by generalized neuromuscular weakness. The reader is referred to other textbooks for further information on these syndromes that are not characterized by seizures.
Diagnosis
Clinical diagnosis is based on history of ingestion, clinical presentation and improvement or resolution of muscarinic signs after atropine administration. If the diagnosis is uncertain, a test dose of atropine (0.02 mg/kg IV) can be administered after evaluating the baseline heart rate. If the heart rate increases, the pupils dilate and hypersalivation stops in 10–15 min, the animal is unlikely to have organophosphate or carbamate intoxication as it takes approximately 10 times this test dose to resolve clinical signs caused by these compounds.
Laboratory findings of heparinized whole blood cholinesterase (ChE) activity reduced by 50% of normal (based on the normal range for that laboratory) suggest exposure, whereas ChE activity less than 25% of normal indicates toxicosis in animals with characteristic clinical signs (Wismer, 2012). ChE activity of heparinized whole blood is a combination of true AChE activity of RBCs and pseudo-ChE activity of serum. Packed cell volume should be checked on blood samples for AChE testing as anaemia can result in decreased AChE activity. Blood should be kept refrigerated to prevent the loss of enzyme activity. ChE activity can remain decreased for 6 to 8 weeks following organophosphate exposure, whereas it may be normal in animals with carbamate intoxication. Carbamates bind reversibly with AChE in the body and they may also dissociate from AChE or pseudo-ChE in a blood tube or other specimen during transit. Stomach content, vomitus, hair, or suspected baits can be submitted to the laboratory for an organophosphate or carbamate residue screen.
Management
Treatment includes administration of atropine
(0.2 to 0.4 mg/kg, one-fourth of the initial dose slowly IV and the rest IM or SQ) to counteract the muscarinic signs, decontamination and prevention of further toxin adsorption (Table 4.1), skeletal muscle relaxants (Table 4.1), AEMs (see Table 4.1 and Chapters 12 and 24) and supportive care. Dramatic cessation of parasympathetic signs is usually observed within 3 to 5 min after administration of atropine IV. Repeated administration of atropine, IV, IM or SC, at one-half of the initial dose is often required, especially in cats with organophosphate toxicity. Glycopyrrolate (0.01–0.02 mg/ kg IV) may also be used to control the muscarinic signs. Cardiac activity should be monitored. In animals with dermal exposure, decontamination is performed by bathing with a mild hand-dishwashing detergent and water. The individuals bathing the animals should wear protective gloves and aprons. In animals with oral exposure, decontamination is performed by induction of emesis (in asymptomatic animals) or gastric lavage and administration of activated charcoal with a saline cathartic or sorbitol (Table 4.1). Fluid therapy should be performed to correct possible dehydration, electrolyte imbalances and acidosis. Endotracheal intubation and ventilation may be necessary in cases of respiratory paralysis.
Pralidoxime chloride (2-PAM) is an AChEreactivating oxime that acts specifically on the organophosphate-enzyme complex and counteracts the nicotinic cholinergic signs (Clemmons, 1990). Administration of 2-PAM should begin within 24 to 48 h of organophosphate intoxication as after this time the toxic compounds are irreversibly bound to AChE. The recommended dose is 10–20 mg/kg slowly IV with fluids over 30 min; or IM or SC. If nicotinic signs persist, the same dose can be repeated every 8–12 h, for 24 to 48 h. 2-PAM should be discontinued after three or four treatments if there is no response or nicotinic signs worsen. 2-PAM produces better results when atropine has already been administered; the atropine dose can be reduced when 2-PAM is used. Signs of muscle weakness and fasciculations usually disappear within 30 min. 2-PAM is not beneficial in treating carbamate toxicosis.
If it is uncertain whether the toxicant is an organophosphate or a carbamate, 2-PAM should be used unless it is likely that the toxicant is carbaryl, in which case 2-PAM may be harmful. Administration of diphenhydramine (1 to 4 mg/kg PO every 8 h) has been recommended by one author to counteract the nicotinic signs 24 to 48 h after intoxication and to prevent signs of subacute intoxication (Clemmons et al., 1984; Clemmons, 1990). However, its use is controversial and other authors consider diphenhydramine contraindicated in animals with organophosphate and carbamate intoxication (Ellenhorn et al., 1997).
Prognosis
Prognosis depends on the dose and duration of exposure to the insecticide and promptness of adequate treatment (Blodgett, 2006). Generally, prognosis is considered good unless the animal shows signs of respiratory dysfunction or seizures (Wismer and Means, 2012).
Chlorinated hydrocarbons
Overview
Chlorinated hydrocarbons, also named organochlorines, have been used for prevention and control of insect infestations around farms, homes and on animals from the 1950s through the 1970s. Chlorinated hydrocarbons include endrin, aldrin, dieldrin, heptachlor, lindane, DDT and endosulfane. Most of these insecticides have been banned because of accumulating tissue residues and environmental persistence. Contaminated soils or leakage from old dump sites are possible sources of exposure for wildlife and domestic carnivores. The most likely source of exposure in dogs and cats is old stockpiles of insecticides and improper waste disposal. In addition, a few of these compounds may still be legal for ectoparasite control in dogs in certain countries. Exposure in dogs and cats may occur by ingestion, inhalation (less likely), or cutaneous absorption when the insecticide is applied topically and accidentally overdosed (Raisbeck, 2006).
Mechanism of action
The mechanism of action of most chlorinated hydrocarbons is poorly understood and they are considered to be nonspecific stimulants of the central nervous system (Hatch, 1988). Persistent opening of neuronal sodium channels and GABA inhibition are possible mechanisms.
Clinical presentation
Clinical diagnosis is based on history of exposure and clinical presentation. Clinical signs include hypersensitivity, nervousness, muscle tremors, spastic gait, ataxia, mydriasis, salivation, vomiting and severe generalized tonic-clonic seizures, which may be precipitated by external stimuli and may last for 2–3 days. Hyperthermia occurs as a result of the seizures. Death may occur within minutes or hours or after several days.
Diagnosis
Confirming the diagnosis is quite difficult as chlorinated hydrocarbon residues may be found in blood or tissue of normal animals due their persistence in tissues (particularly fat) (Raisbeck, 2006). Tissue samples should be submitted in glass or metal containers rather than plastic ones.
Management
Treatment is symptomatic since there is no known antidote and involves dermal
(e.g. bathing) or gastrointestinal (e.g. emesis or gastric lavage, repeated administrations of activated charcoal due to enterohepatic recirculation) (Table 4.1) decontamination, skeletal muscle relaxants (Table 4.1), AEMs (see Table 4.1 and Chapters 12 and 24) and supportive care (including ventilator support in severely affected animals). Forced diuresis with 5% mannitol in 0.9% sodium chloride can enhance urinary excretion.
Prognosis
Signs of acute toxicosis usually abate in 1 to 2 days. Complete recovery may take weeks. The prognosis is guarded to good, depending on the dose of exposure, severity of neurological dysfunction and promptness of treatment.
Molluscicides
Metaldehyde
Overview
Metaldehyde is a cyclic tetramer of acetaldehyde included in a variety of snail and slug baits, most commonly in the form of green granules, but also as liquid, powder or pellets (Yas-Natan et al., 2007).
Protein-rich material, such as bran or grain, is usually added to the bait to make it more attractive to slugs and snails, causing this type of bait to be palatable to dogs as well. Baits are sometimes mixed with other pesticides, most commonly with carbamate insecticides. In some countries, metaldehyde is also used as a fuel in small heating systems, such as camping stoves and lamps. Metaldehyde is degraded to various aldehydes in the stomach resulting in a formaldehyde odour of the gastric contents.
Mechanism of action
The exact mechanism of metaldehyde toxicity is currently unclear and may be associated with an increase in monoamine oxidase activity and a decrease in gamma-aminobutyric acid (GABA), norepinephrine and serotonin concentrations.
Clinical presentation
Clinical signs occur within 20 min to 3 h from ingestion and include tachycardia, tachypnea, hypersalivation, muscle tremors, vomiting, hyperesthesia, nystagmus (especially in cats), ataxia, opisthotonus, seizures, hyperthermia, diarrhoea and obtunded mental status to coma (Yas-Natan et al., 2007). The seizures are tonic, similar to those secondary to strychnine intoxication, but generally they do not worsen with stimuli in dogs. However, in cats seizures have been reported to be triggered or exacerbated by auditory, visual or tactile stimuli. Metabolic acidosis is common. Death may occur from respiratory failure 4 to 24 h after ingestion or from liver failure 3 to 4 days after ingestion in dogs. Post-metaldehyde intoxication liver disease has not been reported in cats.
Diagnosis
Diagnosis can be reached by laboratory analysis of gastric contents, serum, urine and liver. Samples must be kept frozen for analysis.
Management
Treatment includes induction of emesis (in mildly affected animals with no seizures and toxin ingestion <3 h), gastric lavage (toxin ingestion >3 h, ingestion of large volumes), activated charcoal (Table 4.1), methocarbamol for muscle tremors, AEMs (see Table 4.1 and Chapters 12 and 24), fluid therapy to correct metabolic acidosis, convective whole body cooling (e.g. wetting the fur, fan) and respiratory support. General anaesthesia may be necessary in animals that do not respond to AEMs (see Chapter 24). After the acute clinical signs have been controlled, treatment must focus on minimizing possible liver damage in dogs.
Prognosis
Prognosis is generally good in animals treated promptly and aggressively. A retrospective study including 18 dogs with metaldehyde intoxication reported a recovery rate of 83% (Yas-Natan et al., 2007). Prognosis is guarded in animals presenting with severe hyperthermia (>41.6°C or 107°F). Prolonged status epilepticus following metaldehyde intoxication has not been associated with spontaneous recurrent seizures in dogs (Jull et al., 2011). Recovery may take approximately 2 weeks in cats (Puschner, 2006).
Rodenticides
Strychnine
Overview
Strychnine has been used as pest control of rodents and other animal species worldwide.
Its sale has been restricted or banned without special permits in several countries. However, malicious and less commonly accidental or secondary (from ingestion of strychnine-poisoned rodents) poisonings of companion animals still occurs (Berny et al., 2010). The toxic dose in most animals ranges from 0.3 to
1.0 mg/kg with the lethal dose being 2.0 mg/kg in cats.
Mechanism of action
Strychnine antagonizes stereochemically and competitively the motor inhibitory neurotransmitter glycine at the brainstem and spinal cord level and inhibits glycine release from Renshaw cells. Some supraspinal signs may also be associated with strychnine inhibition of GABA.
Clinical presentation
Clinical signs generally occur within 10 min to 2 h after toxin ingestion and typically begin with nervousness and restlessness, rapidly progressing to increased tone of both extensor and flexor muscles resulting in a stiff gait. All skeletal muscles including the appendicular, epaxial, facial, abdominal and respiratory muscles have tetanic spasms. Auditory
(e.g. loud noise), visual (e.g. bright light) and tactile stimuli exacerbate the tetanic muscle spasms and can trigger tonic seizures. This feature, however, is not specific to strychnine and has been observed in other types of poisonings including metaldehyde, penitrem A, roquefortine and chlorinated hydrocarbons. Hyperthermia (secondary to the severe muscle contractions) is commonly observed. The animal remains conscious during seizures unless respiratory paralysis occurs. Apnea can lead to cerebral anoxia, loss of consciousness and death 30 min to 2 h following the onset of neurologic signs, if the animal is untreated (Murphy, 2002).
Diagnosis
Baits may have specific colours in some countries, which can help recognition during gastric decontamination. Definitive diagnosis is based on toxicologic analysis of stomach contents, urine, blood or hepatic and renal tissue. Ante-mortem analysis of stomach contents and urine is most likely to provide a diagnosis. The dimethoxy derivative of strychnine, brucine (2,3-dimethoxystrychnidin-10-one), may be detected in serum.
Management
Treatment includes decontamination (induction of emesis in asymptomatic animals or gastric lavage followed by activated charcoal and cathartic administration in animals showing clinical signs of intoxication) (Table 4.1), promoting toxin excretion, controlling the tetany and seizures (Table 4.1, see Chapters 12 and 24), convective whole body cooling, adequate oxygenation (Murphy, 2002) and supportive care. Sedation or general anaesthesia for 24–72 h may be required. Forced diuresis with 5% mannitol in isotonic saline and acidification of the urine will enhance urinary elimination of strychnine. Animals with respiratory failure should be administered oxygen and if needed intubated and ventilated. Non-anesthetized animals should be kept in a dimly lit, quiet area and all forms of sensory stimulation should be minimized.
Prognosis
Prognosis is fair to guarded, depending on the amount of toxin ingested and promptness of treatment. If the animal survives the first 24 h post-toxin ingestion, prognosis for complete recovery is good.
Bromethalin
Overview
Bromethalin is a rodenticide that is sometimes implicated in accidental or malicious poisonings of small animals. It is available in pelleted forms such as place packs, blocks or bars of bait and baited worms (DeClementi and Sobczak, 2012). Exposure mainly occurs by ingestion of bromethalin bites. Secondary (or relay) poisoning may occasionally occur by ingestion of bromethalin-poisoned rodents. Bromethalin is readily absorbed from the gastrointestinal tract and reaches peak plasma levels within several hours after ingestion.
It is highly lipophilic and therefore reaches high concentration in the brain.
Mechanism of action
Bromethalin and its active metabolite, desmethyl bromethalin, uncouple oxidative phosphorylation resulting in decreased cellular adenosine triphosphate (ATP) concentrations and reduced activity of ATP-dependent sodium and potassium ion channel pumps. This produces an increase in intracellular sodium concentrations, intracellular movement of water and consequent cerebral oedema, vacuolization of myelin and increased intracranial pressure.
Clinical presentation
Onset and type or clinical signs are dose-dependent. Dogs ingesting 2.5 to 5 mg/kg of bromethalin develop clinical signs in 1 to 4 days (Murphy, 2002), whereas dogs ingesting more than 5 mg/kg of bromethalin may develop clinical signs within 2–24 h after ingestion. Low doses (e.g. 2.5 mg/kg in dogs) result in pelvic limb ataxia and/or paresis/paralysis with extensor rigidity and obtunded mental status. High doses (e.g. 5 to 6.5 mg/kg in dogs) produce hyperexcitability, tremors, focal motor and generalized seizures, hyperthermia, obtunded mental status, which can progress to stupor, coma, decerebrate posture and death secondary to respiratory arrest (Dorman et al., 1990a, b). Cats ingesting 0.54 mg/kg develop signs (including ataxia, focal motor seizures, vocalization, obtunded mental status and stupor) 2 to 7 days after exposure (Dorman et al., 1990c). Most recently clinical signs and death have been reported following exposure to bromethalin doses as low as 0.46 mg/kg in dogs and 0.24 mg/kg in cats (DeClementi and Sobczak, 2012).
Diagnosis
The ante-mortem diagnosis is most often made based on the history of bait ingestion (stools may have a green discoloration) and the development of clinical signs. Definitive diagnosis can be reached only post-mortem by detecting bromethalin or its metabolites in kidney, liver, fat or brain samples using gas chromatography with electron capture (Dorman et al., 1990a, b). Samples should be submitted frozen and protected from light.
Management
There is no antidote to bromethalin. Treatment involves decontamination and prevention of further toxin adsorption (induction of emesis or gastric lavage followed by repeated administration of activated charcoal and administration of a cathartic with the initial dose of activated charcoal) (Table 4.1), AEMs (see Table 4.1 and Chapters 12 and 24) in seizuring animals, methocarbamol or diazepam in animals with excessive tremors (Table 4.1), and supportive care. The repeated and prolonged administration of activated charcoal (1–5 g/kg every 6–8 h for up to 2–4 days in animals ingesting high dosage of bromethalin) is indicated due to the enterohepatic recirculation of bromethalin. Cerebral oedema can be treated with mannitol. Serum sodium levels should be closely monitored due to the potential for hypernatraemia of repeated administration of activated charcoal as well as mannitol.
Prognosis
Prognosis is fair with prompt and prolonged treatment in animals with mild clinical signs. Animals with obtundation and ataxia may recover over a period of 2 to 4 weeks. Prognosis is guarded in animals with severe neurological signs such as tremors, seizures, coma or paralysis.
Zinc phosphide
Overview
As with other rodenticides, zinc phosphide poisoning can be accidental or malicious in small animals. Secondary (or relay) poisoning has been reported in dogs feeding on the dead rodents and other animals poisoned by zinc phosphide. When zinc phosphide reaches the stomach, on exposure to moisture and an acidic environment, it hydrolyses to phosphine gas which is rapidly absorbed across the gastric mucosa and distributed systemically where it exerts its toxic effect (Proudfoot, 2009).
Mechanism of action
The corrosive action of zinc phosphide accounts for the early, acute and generally haemorrhagic emetic effect on the gastric mucosa. The systemic toxicity of zinc phosphide is caused by phosphine. Postulated mechanisms of action of phosphine include inhibition of cytochrome C oxidase with mitochondrial dysfunction and interruption of cellular respiration, inhibition of serum acetyl cholinesterase activity resulting in cholinergic overdrive and formation of reactive oxygen species (ROS) with resultant oxidative stress, damage to cell lipids, proteins and nucleic acids and cell death (Proudfoot, 2009).
Clinical presentation
Clinical signs occur within 15 min to 4 h after ingestion (but may be delayed by 12 to 18 h) and include vomiting (often with frank or dark blood clots), aimless pacing or running, vocalization, anxiety, discomfort, generalized muscle fasciculations, tremors, exaggerated response to external stimuli and tonic-clonic or tonic seizures. In addition, the production of phosphine gas within the stomach may lead to gastric or abdominal distension resulting in abdominal pain and potentially gastric dilatation-volvulus. Death can occur (Murphy, 2002; Proudfoot, 2009).
Diagnosis
The odour of phosphine is similar to rotten fish or acetylene gas and may be detected on the breath or in the stomach contents of intoxicated animals. However, some types of zinc phosphide may also be odourless. Owners should be warned about the risk of phosphine gas inhalational exposure during transit to the veterinary hospital and adequate precautions should be taken to minimize veterinary and support personnel exposure. The presence of zinc phosphide (phosphine gas) can be detected in stomach contents, vomitus, or suspect bait by laboratory analysis (gas chromatography-mass spectrometry or Dräger detector tube test) (Murphy, 2002; Proudfoot, 2009). The sample should be frozen in an airtight container soon after collection to prevent loss of phosphine gas.
Management
No specific antidote exists. Treatment involves decontamination (gastric lavage and activated charcoal), reducing phosphine production by decreasing the acidity of the gastric lumen through administration of a liquid antacid
(e.g. magnesium or aluminium hydroxide and calcium carbonate) or 2–5% solution of sodium bicarbonate orally or through gastric lavage tube, symptomatic and supportive care (intravenous fluids, oxygen supplementation, gastro protectants, analgesia and hepatic supportive agents) and AEMs (see Table 4.1 and Chapters 12 and 24). Adequate room ventilation is imperative during gastrointenstinal decontamination (Gray et al., 2011).
Prognosis
Prognosis can be favourable in animals treated promptly and effectively (Murphy, 2002; Gray et al., 2011).
Sodium monofluoroacetate (Compound 1080)
Overview
Sodium monofluoroacetate was introduced as a rodenticide in the USA in 1946 and subsequently used as pest control particularly of non-native species such as the fox and possum in Australia and New Zealand, respectively. Sodium monofluoroacetate is one of the most toxic pesticides and its use is restricted to trained, licensed applicators. However, accidental or malicious poisoning of domestic animals can occur in baiting areas. Accidental or malicious poisoning of companion animals can occur directly from bait ingestion or secondarily following the ingestion of a poisoned carcass. Sodium monofluoroacetate can be absorbed from the gastrointestinal and respiratory tracts as well as across mucous membranes and abraded skin. Dogs are more susceptible than cats to this toxicant.
with oxaloacetate to produce fluorocitrate. Fluorocitrate is converted to 4-hydroxytrans-aconitate, which binds and inactivates aconitase resulting in inhibition of citrate oxidation. This results in inhibition of the tricarboxylic acid (TCA) or Kreb’s cycle, cellular energy depletion, citric acid and lactic acid accumulation, a decrease in blood pH, and interference with cellular respiration and metabolism of carbohydrates, lipids and proteins. Organs with cells with a high metabolic rate, such as the heart, brain and kidneys, are most susceptible to dysfunction (Goh et al., 2005; Parton, 2006; Proudfoot et al., 2006). In addition to blockade of the TCA cycle, citrate accumulates within blood to toxic concentrations and binds to calcium resulting in serum ionized hypocalcaemia.
Clinical presentation
Clinical signs occur 30 min to 2 h after ingestion, depending on the dose, and include restlessness, hyperirritability, hyperesthesia, running, barking and howling episodes, vomiting, salivation, defecation, diarrhoea, urination, tremors, tonic-clonic seizures, hyperthermia (in dogs) and eventually coma and death 2 to 12 h after the onset of clinical signs. Hypothermia, vocalization, cardiac arrhythmias and episodes of bradycardia between seizures have been reported in cats.
Diagnosis
Diagnosis of sodium monofluoroacetate poisoning is usually based on characteristic clinical signs in conjunction with known access to the poison. Clinical pathological changes include metabolic acidosis, serum-ionized hypocalcaemia and elevations in serum citrate concentrations over two to three times greater than the reference range. Analysis of gastric content from vomitus or lavage fluids can confirm the diagnosis. The sample should be kept frozen until analysis to avoid bacterial breakdown of the toxin.
Mechanism of action Management
Fluoroacetate combines with acetyl-CoA to Treatment involves decontamination (inducform fluoroacetyl-CoA, which then combines tion of emesis in asymptomatic animals or
gastric and colonic lavage in symptomatic animals, administration of activated charcoal with a cathartic) (Table 4.1), methocarbamol or diazepam in animals with excessive tremors (Table 4.1), AEMs (see Table 4.1 and Chapters 12 and 24) in seizuring animals, symptomatic and supportive care. Calcium gluconate (see hypocalcaemia) should be administered if serum-ionized calcium concentration is equal or lower than 0.8 mmol/l (3.2 mg/dl). In addition, administration of sodium bicarbonate or acetamide has shown promising results (O’Hagan, 2004; Parton, 2006). The recommended dosage of sodium bicarbonate is 300 mg/kg (3.6 ml/kg of 8.4% solution) IV over 15–30 min. Alternatively, half of the calculated dose may be given as a bolus and the remainder infused slowly. Administration of sodium bicarbonate may worsen hypocalcaemia and cause hypokalaemia and hypernatraemia. Serum-ionized calcium, sodium and potassium levels should be monitored regularly in order to implement fluid therapy as well as calcium and potassium supplementations as required.
The recommended dosage of acetamide (15 g of acetamide granules dissolved in 1 l of warmed 5% dextrose) in dogs is 10 to 25 ml/kg IV (infused though a filter following sterilization) over a 60-min period followed by approximately 5 ml/kg/h IV for the next 12 to 18 h until resolution of clinical signs. In cats with fluoroacetate poisoning, the acetamide dose should be reduced by at least 75% (Parton, 2006). Electrolytes should be closely monitored as hyponatraemia may develop due to the large volume of administered free water.
Other treatment modalities are being investigated in Australia and New Zealand due to the higher prevalence of sodium monofluoroacetate poisoning than other countries.
Prognosis
The prognosis is poor to grave, depending on the amount of sodium monofluoroacetate ingested and the severity of clinical signs at initial evaluation (Goh et al., 2005). Early acetamide or sodium bicarbonate treatment and good supportive care can improve survival (O’Hagan, 2004; Parton, 2006).
Automotive Products
Ethylene glycol
Overview
Ethylene glycol is a commercial antifreeze automotive product whose metabolites, including glycolic acid, are extremely toxic to dogs and cats. Common sources of exposure include container spill, engine flush or engine leak (Khan et al., 1999). Intoxication is most commonly accidental but can also be malicious.
Mechanism of action
Ethylene glycol is biotransformed to glycolic acid, which is metabolized to formic acid, oxalic acid and oxalate. These metabolites are highly toxic and result in severe metabolic acidosis and acute renal failure. The oxalate combines with calcium to form oxalate crystals in renal tubules (especially proximal), urine and within the lumen or perivascular space of cerebral capillaries (Dial et al., 1994a).
Hypocalcaemia secondary to calcium oxalate deposition may contribute to CNS signs, although the concurrent metabolic acidosis shifts calcium to the ionized active state, reducing the chances of hypocalcaemia-associated clinical signs. Acidosis may also contribute to cerebral damage.
Clinical presentation
Clinical signs usually occur within 30 to 60 min of exposure and include obtundation, vomiting, ataxia, seizures, hypothermia, severe metabolic acidosis, serum hyperosmolality, polydipsia, polyuria, calcium oxalate monohydrate and dihydrate crystalluria, isosthenuria and eventually renal failure (Dial et al., 1994a).
Diagnosis
Ethylene glycol colorimetric spot tests are available for use with urine and serum. However, these tests can give false negatives in cats as the ethylene glycol toxic dose in cats can be below the detectable level of the
ethylene glycol test kit. False positive results Management
can occur in animals administered medications containing propylene glycol (diazepam, activated charcoal). A quantitative test kit has recently been evaluated and may aid in timely diagnosis of ethylene glycol exposure (Scherk et al., 2013). Laboratory tests for rapid analysis of serum, plasma or urine for ethylene glycol and glycolic acid also have been reported (Smith and Lang, 2000; Van Hee et al., 2004). Birefringent crystals may be detected in urine 3 h (in cats) and 5 h (in dogs) after ingestion. Anion gaps greater than 40–50 mEq/l are also suggestive of ethylene glycol intoxication. Signs of acute renal failure (azotaemia, hyperkalaemia, hyperphosphataemia) are usually seen approximately 12–48 h post-ethylene glycol ingestion. Moderate to severe hypocalcaemia is frequently present. Serum osmolality as high as 450 mosm/kg serum and an osmole gap as high as 150 mosm/kg serum may be detected 3 h after ethylene glycol ingestion. Both the gap and the measured osmolality may remain elevated for approximately 18 h after ingestion. Ultrasonographic changes vary from mild to marked increases in renal cortical echogenicity. Another diagnostic procedure that may help in the early diagnosis of ethylene glycol intoxication is examination of the oral cavity, face, paws, vomitus and urine with a Wood’s lamp to determine whether they appear fluorescent. Many antifreeze solutions contain sodium fluorescein, a fluorescent dye that aids in the detection of leaks in vehicle coolant systems (Thrall et al., 2006).
Treatment should be instituted promptly even if the results of confirmatory tests are not available yet. Treatment is aimed at preventing absorption, increasing excretion and preventing metabolism of ethylene glycol using a chemical antidote such as fomepizole or ethanol. Although therapeutic recommendations have traditionally included induction of vomiting, gastric lavage and administration of activated charcoal, it is likely that these procedures are not beneficial because of the rapidity of ethylene glycol absorption (Thrall et al., 2006). In addition, absorption of ethanol is inhibited by charcoal. Fomepizole (4-methylpyrazole), a competitive inhibitor of alcohol dehydrogenase, is considered safe and effective for dogs if started within 8 h of exposure (Table 4.2) (Dial et al., 1994a; Connally et al., 1996). Fomepizole can be used also in cats, although a much higher dosage is required (Dial et al., 1994b). If Fomepizole is not available, 20% ethanol can be used (Table 4.2). It acts as a competitive substrate for the enzyme alcohol dehydrogenase. Although effective in the treatment of ethylene glycol toxicity, ethanol may result in CNS and respiratory depression. Supportive and symptomatic care includes intravenous fluids to correct dehydration, acid-base and electrolyte imbalances and to promote diuresis, and AEMs in seizuring animals (see Table 4.1 and Chapters 12 and 24).
Prognosis
The mortality rate in dogs is reported to range from 59% to 70% and is thought to be even
Table 4.2. Fomepizole and ethanol dosage in the treatment of ethylene glycol toxicity.
Medication Species Dosage
| Fomepizole | Dog | 20 mg/kg IV initially as a loading dose, followed by 15 mg/kg IV at 12 and 24 h, |
| and 5 mg/kg IV at 36 h | ||
| Fomepizole | Cat | 125 mg/kg IV initially, followed by 31.25 mg/kg IV 12, 24 and 36 h after the |
| initial bolus | ||
| 20% ethanol | Dog | 5.5 ml/kg of a 20% solution IV every 4 h for five treatments, then every 6 h for |
| four additional treatments; or 22 ml/kg IV of a 5% solution every 4 h for six | ||
| treatments, then every 6 h for four treatments; or a constant rate IV infusion | ||
| of 5% solution at 5.5 ml/kg/h | ||
| 20% ethanol | Cat | 5 ml/kg of a 20% solution IV every 6 h for five treatments, then every 8 h for four |
| additional treatments |
higher in cats. However, prognosis has been reported to be favourable in dogs and cats treated within 8 and 3 h following ingestion, respectively (Thrall et al., 2006).
Detergents and Disinfectants
Hexachlorophene
Overview
Hexachlorophene is used as a germicide in soaps, shampoos and disinfectant solutions. Exposure may result from both topical contact and ingestion (Bath, 1978; Thompson et al., 1987). Nursing puppies have been poisoned following hexachlorophene application to the mammary glands of the bitch (Ward et al., 1973).
Clinical presentation
Clinical signs in dogs are usually characterized by acute onset of tremors, especially of the head, which may increase with excitement and disappear during rest or sleep. Opisthotonus, severe seizures and death have also been reported. Clinical signs in cats include mydriasis, vomiting, weakness, ataxia, spastic or flaccid paralysis and hypovolaemic shock (Thompson et al., 1987).
Management
Treatment involves decontamination, supportive care, skeletal muscle relaxants and AEMs (see Table 4.1 and Chapters 12 and 24).
Prognosis
Clinical recovery may take 4 to 6 weeks.
Heavy Metals
Lead
Overview
Lead is the most common heavy metal causing toxicosis in animals. Sources of lead intoxication include lead-based paints, linoleum, putty, roofing felt, golf balls, bullets or pellets, fishing weights, old car batteries, wheel weights, improperly glazed ceramic water bowls and toys (Morgan, 1994). The most commonly identified source of exposure in dogs and cats is lead-based paint chips or dust, usually from home renovation. Cats can ingest old paint dust and chips contaminating the coat while grooming (Knight and Kumar, 2003). High-fat low-calcium diets may facilitate absorption of lead from the alimentary tract.
Mechanism of action
The toxic mechanism of lead is caused predominantly by its ability to substitute for other polyvalent cations (particularly divalent cations, such as calcium (Ca2+) and zinc (Zn2+) ) in multiple molecular processes, and subsequently affect metal transport, energy metabolism, membrane ionic channel conduction, inter- and intracellular signalling, diverse enzymatic processes (including sulfhydryl-containing enzymes involved in haem synthesis), protein maturation, cell adhesion and regulation of gene transcription. The effects of lead neurotoxicity include decreased cellular energy metabolism, impaired haem biosynthesis (resulting in increased erythrocytes fragility, basophilic stippling and circulating nucleated erythrocytes), oxidative stress, lipid peroxidation, altered activity of second messenger systems, altered neurotransmitter release (e.g. decreased GABA neurotransmission) and neurotransmitter receptor density, excitotoxicity, apoptosis impaired development and function of oligodendrocytes, abnormal myelin formation, abnormal neurotrophic factor expression, abnormal dendritic branching patterns and disruption of the blood-brain barrier (Lidsky and Scneider, 2003; Garza et al., 2006).
Clinical presentation
Clinical presentation is variable depending on duration and degree of exposure. Gastrointestinal signs (including vomiting, anorexia, diarrhoea and sometimes also abdominal pain) precede or accompany neurological signs (including seizures, blindness, obtundation, hyperexcitability, behavioural changes, head pressing, ataxia and tremors) (Zook et al., 1967; Knecht et al., 1979; Morgan, 1994). Clinical signs in cats may be more vague than in dogs and most commonly include anorexia, vomiting and seizures. Central vestibular abnormalities, including vertical nystagmus and ataxia, have also been reported in cats with lead poisoning (Knight et al., 2001). Clinical signs can be acute or chronic. In cases of chronic exposure,the seizures are intermittent(Bratton and Kowalczyk, 1989).
Diagnosis
Haematology and serum biochemistry may reveal one or more of the following abnormalities: nucleated erythrocytes and basophilic stippling in red blood cells with no or mild anaemia, increased packed cell volume, leukocytosis, elevated hepatic enzymes, hyperglycaemia and hypercholesterolaemia. Radiography may allow identification of metallic material in the gastrointestinal tract (e.g. golf balls or toys) or subcutaneously (e.g. pellets). Blood lead levels of 40 mg/dl or higher are considered diagnostic of lead poisoning (Morgan, 1994). However, blood lead concentration fluctuates and due to sequestration in other organs does not necessarily correlate with total body burden of lead or with clinical signs (Bratton and Kowalczyk, 1989; Knight and Kumar, 2003). If the blood lead values are in the high normal range and lead poisoning is suspected clinically, treatment followed by measurement of urine lead levels can be diagnostic. Electroencephalographic changes in non-sedated dogs are characterized by intermittent high-amplitude slow wave activity (Knecht et al., 1979). A post-mortem diagnosis can be made by analysing lead concentration in kidneys, liver, brain and bones (Knight and Kumar, 2003). Levels of 3.6 to 10 mg/g by wet weight of liver tissue are considered diagnostic of lead toxicosis (Bratton and Kowalczyk, 1989).
Management
Treatment of lead toxicosis involves prevention of further exposure, decontamination of the individual (e.g. bathing in case of lead-laden dust coat exposure, administration of cathartics and/or enemas, endoscopic or surgical removal of lead-containing foreign bodies), decontamination of the environment, supportive care and chelation therapy to remove lead from the blood and soft tissues. The cathartic magnesium sulfate (250–500 mg/kg PO in dogs and 200 mg/kg PO in cats) can help to decrease lead absorption by forming insoluble lead sulfate. Treatment with the chelating agent, calcium disodium ethylene diamine tetraacetate (CaNa2EDTA), using a dose of 25 mg/kg intravenously or orally, four times a day for 2 to 5 days, has resulted in recovery within 36 to 48 h. However, in some animals, CaNa2EDTA has initially worsened neurologic signs. The most recently available chelator, succimer (meso-2,3-dimercaptosuccinic acid), administered at 10 mg/kg of body weight, orally, every 8 h, for 10 days, has been reported to be safe and effective in the treatment of lead poisoning in dogs and cats (Ramsey et al., 1996; Knight et al., 2001). Succimer may also be administered rectally as a solution in patients that are vomiting or that are unable to take oral medications. Chelation therapy with calcium EDTA (27 mg/kg SC for 5 days) alone or in association with D-penicillamine (33–55 mg/kg/day divided every 6–8 h) has also been reported. Seizures should be treated promptly with diazepam and/or other AEMs (see Table 4.1, and Chapters 12 and 24). Thiamine supplementation (1–2 mg/kg IM or 2 mg/kg PO every 24 h) can contribute to neurological improvement.
Prognosis
Clinical response to therapy is the best prognostic indicator. Prognosis is favourable in the majority of lead-poisoning cases treated with chelating agents (Morgan, 1994). Continuous or uncontrolled seizures may be associated with a less favourable prognosis.
Poisonous Plants
Seizures have been reported following exposure to the plants listed in Box 4.7 (Barr, 2006).
Several of these plants cause vomiting. The gastric content from vomitus or lavage fluids can be submitted for plant material
Box 4.7. Poisonous plants.
Amaryllis (Amaryllis spp.) Angel’s trumpet (Brugmansia spp.) Aralia, balfour aralia, dinner plate aralia, ming
aralia, geranium-leaf aralia, wild coffee, coffee tree (Polyscias spp.) Black locust (Robinia pseudoacacia)
Bleeding heart, Dutchman’s breeches, squirrel
corn, staggerweed (Dicentra spp.) Box, common box, boxwood (Buxus sempervirens) Broom (Cytisus spp.)
Candelabra cactus, false cactus, mottled
spurge, dragon bones (Euphorbia lactea) Castor bean (Ricinus communis) Chinaberry tree (Melia azedarach) Croton (Croton tiglium) English ivy, Irish ivy, common ivy (Hedera helix) Euphorbium (Euphorbia resinifera) Golden chain tree (Laburnum anagyroides) Golden corydalis, bulbous corydalis, scrambled
eggs, fitweed, fumitory (Corydalis spp.) Meadow saffron, autumn crocus (Colchicum autumnale) Mediterranean thistle (Atractylis gummifera) Milkweed, butterfly weed (Asclepias spp.) Mimosa, silk tree (Albizia julibrissin)
Pencil tree, milkbush, Indian tree, rubber euphorbia,
finger tree, naked lady (Euphorbia tirucalli)
Persian violet, alpine violet, sowbread
(Cyclamen spp.)
Sago palm, leatherleaf palm, Japanese fern
palm (Cycas revoluta, Cycas spp.) Sandbox tree, monkey pistol (Hura crepitans) Schefflera, umbrella tree, rubber tree, starleaf
(Schefflera spp.) Sea onion (Urginea maritima) Spurge, creeping spurge, donkey tail (Euphorbia myrisinites)
Squill, starry hyacinth, autumn scilla, hyacinth scilla, Cuban lily, Peruvian jacinth, hyacinth of Peru, bluebell (Scilla spp.) Snowdrop (Galanthus nivalis) Tobacco, tree tobacco (Nicotiana spp.) Tung oil tree (Aleurites spp.) Water hemlock, cowbane (Cicuta spp.) White cedar (Thuja occidentalis)
Yellow jessamine, Carolina jessamine
(Gelsemium sempervirens) Yesterday today and tomorrow (Brunfelsia spp.) Zamia (Macrozamia spp.) Zulu potato, climbing onion (Bowiea volubilis)
information on geographic distribution of these plants and clinical presentation, treatment and prognosis following exposure is beyond the scope of this book and can be found elsewhere (Barr, 2006).
Blue-green algae (cyanobacteria)
Overview
Blue-green algae (cyanobacteria) are commonly found growing in fresh and salt water in temperate areas worldwide. Under certain climatic and nutritional conditions cyanobacteria reproduce explosively, resulting in algae blooms that accumulate at the water surface and sometimes produce hepatotoxins and/or neurotoxins. Dogs can be exposed by drinking or ingesting contaminated water while swimming (Edwards et al., 1992; Gugger et al., 2005; Puschner et al., 2010; Elford et al., 2012; Faassen et al., 2012). Cats seem to be affected less commonly than dogs.
Mechanism of action
Blue-green algae Anabaena, Aphanizomenon, Lyngbya and Oscillatoria can produce the neurotoxins anatoxin-a (a potent postsynaptic depolarizing neuromuscular blocking agent) and anatoxin-as (a potent acetylcholinesterase inhibitor) (Hooser and Talcott, 2006).
Clinical presentation
Clinical signs often occur very rapidly (10 min to a few hours) after oral exposure and include salivation, lacrimation, urination and defecation, muscle rigidity, muscle tremors, seizures, appendicular paralysis, respiratory paralysis and death within 30 to 60 min of the onset of clinical signs.
Diagnosis
Confirmation of exposure can be achieved identification. Treatments for toxic plant by detection of the algae in gastric con-ingestion include decontamination and sym-tents or suspect water source and detecptomatic and supportive care (including AEMs) tion of the toxins in stomach contents or (see Table 4.1, Chapters 12 and 24). Detailed liver.
Management
Treatment includes decontamination (bathing if the animal went into the contaminated water, gastric lavage, activated charcoal and/ or a cathartic) (Table 4.1), AEMs in seizuring animals (see Table 4.1 and Chapters 12 and 24) and aggressive supportive care including intravenous fluid therapy, atropine (in animals exposed to anatoxin-as neurotoxin) and respiratory support (Hooser and Talcott, 2006).
Prognosis
The prognosis for animals that show clinical signs is guarded to poor.
Mycotoxins
Penitrem A and roquefortine
Overview
The mycotoxins penitrem A (produced by Penicillium crustorum) and roquefortine (produced predominately by Penicillium roqueforti as well as other Penicillium species including
P. crustorum) can be found in mould-contaminated cheese, bread, rice, walnuts and decaying organic matter such as silage, garbage and compost. Absorption after oral ingestion is rapid. Dogs are more commonly affected than cats, probably because of their tendency to scavenge and ingest rotting material (Barker et al., 2013).
Mechanism of action
The exact mechanism of action of these mycotoxins is unknown; however, results of in vitro studies indicate that penitrem A may antagonize the release or action of glycine in the CNS and interfere with the release of glutamate, aspartate and gamma-aminobutyric acid in central and peripheral synapses (Young et al., 2003).
generalized muscle tremors (generally 2–3 h following ingestion), seizures, ataxia, tachycardia and hyperthermia (Lowes et al., 1992; Young et al., 2003; Eriksen et al., 2010). Seizures can sometimes be triggered by auditory (e.g. loud noise), visual (e.g. bright light) and tactile stimuli. Status epilepticus can occur.
Diagnosis
The presumptive diagnosis is based on the history of ingestion of mouldy food or compost and exclusion of other aetiologies. Definitive diagnosis is made by identification of the mycotoxins by means of laboratory analysis of samples of the ingested material, gastric content, serum or urine. Liquid chromatography-mass spectrometry can be used to screen for the mycotoxins penitrem A and roquefortine in serum and urine samples.
Management
Treatment involves reducing toxin absorption (by induction of emesis and/or gastric lavage and administration of activated charcoal and an osmotic cathartic) (Table 4.1), methocarbamol to control the muscle tremors and AEMs (see Table 4.1 and Chapters 12 and 24) and supportive care (intravenous fluid therapy, convective whole body cooling and, in severely affected animals, intubation and ventilation). Emesis should be induced in asymptomatic animals that present within 15–30 min after suspected or confirmed ingestion of mouldy food or non-corrosive waste material (Barker et al., 2013). Administration of activated charcoal should be repeated over 1–3 days due to entero-hepatic recirculation of mycotoxins. Seizures may not respond to diazepam administration and it has been suggested that poor or no response to diazepam is suggestive of mycotoxicosis (Barker et al., 2013). If there is inadequate response after the first administration of diazepam, alternative AEMs such as levetiracetam, phenobarbital, propofol and ketamine (see Table 4.1 and Chapter 24) should be used.
Clinical presentation Prognosis
Ingestion of contaminated material results in The majority of dogs recover in a few days salivation, vomiting, diarrhoea, restlessness following prompt and appropriate treatment; and panting, generally followed by severe however, severely affected animals may take
L. De Risio
several months to achieve complete resolution of neurological signs (Eriksen, 2010).
Animal-related Poisoning
Neurological signs including seizures can occur following exposure to venom of several animal species including insects (bee, wasp), spiders, reptiles and amphibians.
Toad
Overview
There are more than 200 species of Bufo toads in the world and they all have parotid glands on their dorsum that release toxic substances when the toad is attacked or threatened. These include bufotoxins and bufagenins, bufotenine, dopamine, epinephrine, norepinephrine, serotonin and indolealkylamines. Most small animals do not consume Bufo toads. Toxicosis occurs in animals that masticate or hold the toad instead of just biting and then releasing it. The toxins are rapidly absorbed through the oral mucosa and enter the systemic circulation. The toxins can also be absorbed through the gastric mucosa following ingestion, through open skin wounds and across the conjunctiva (Peterson and Roberts, 2006). Toad intoxication has been diagnosed in dogs and cats, however, dogs are more commonly affected (Roberts et al., 2000). Chances of exposure are higher when toads are most active (i.e. in the evening after high rainfall or with high temperatures).
Mechanism of action
Bufotoxins and bufagenins are digitalis-like compounds, which result in cardiac arrhythmias. In addition, bufotoxins cause vasoconstriction. Bufotenins are hallucinogenic. Epinephrine, norepinephrine and dopamine cause tachycardia, hypertension and seizures.
Clinical presentation
The toxins produced by the Bufo marinus toad (Fig. 4.6) (which is found in Florida, USA and north and north-east Australia) have been reported to result in neurological signs including seizures, ataxia, nystagmus, extensor rigidity, opisthotonos and stupor in dogs (Roberts et al., 2000). The neurological signs occur within minutes to 1 h after exposure and are preceded by oral mucous membrane hyperaemia, hypersalivation and pawing at the mouth. Cardiac arrhythmias often occur. Severity of signs is directly proportional to toxin dose. Death may occur following prolonged seizure activity and cerebral oedema or cardiac arrest.
The European Bufo vulgaris (common toad) and the Asian Bufo gargarizans are related species to the Bufo marinus toad and can also cause poisonings, although seizures have not been described (Peterson et al., 2006).

Fig. 4.6a, b. Bufo marinus toad (courtesy of Courtney Freeman ©). Reactive Seizures
Diagnosis
No true confirmatory tests exist for Bufo toad intoxication. Clinical diagnosis is based on history of exposure and presenting signs. Digoxin serum immunoassays can identify the digitalis-like compounds produced by the toad parotid glands in the serum of the intoxicated animal.
Management
Treatment involves decontamination by thorough flushing of the oral cavity with running water for at least 5 min, AEMs (see Table 4.1; Chapters 12 and 24), detection and management of cardiac arrhythmias, intravenous fluids and diuretics to promote urinary excretion of the toxin. Emesis, gastric lavage or endoscopic removal is indicated in the rare event of toad ingestion.
Prognosis
Overall mortality is low in animals treated within a few hours of exposure (Roberts et al., 2000; Reeves, 2004).
Therapeutic Agents and Supplements
Metronidazole
Overview
Metronidazole is a nitroimidazole antibacterial and antiprotozoal agent used in the treatment of giardiasis, anaerobic infections and inflammatory bowel disease (Evans et al., 2003). Neurotoxicity from metronidazole has been reported in dogs receiving as low as 67 mg/ kg/day for an average of 3–14 days (Dow, 1989) or 60.3 mg/kg/day for 44.9 days (Evans et al., 2003), and in cats receiving 111 mg/kg/ day for 9 weeks or 58 mg/kg/day for 6 months (Caylor and Cassimatis, 2001).
Mechanism of action
The neurotoxic mechanism of metronidazole has not been identified. Proposed mechanisms include thiamine antagonism and inhibition of neuronal protein synthesis.
Clinical presentation
Neurological signs in cats include disorientation, ataxia, seizures and blindness (Caylor and Cassimatis, 2001). Neurological signs in dogs are characterized by cerebellar and central vestibular dysfunction (Dow et al., 1989; Evans et al., 2003). Neurologic signs may be preceded or associated with anorexia, vomiting and/or diarrhoea.
Diagnosis
Diagnosis is based on the history of metronidazole administration, clinical signs and resolution of signs following metronidazole discontinuation.
Management
Treatment involves metronidazole discontinuation, symptomatic and supportive care. In dogs, diazepam has been reported to markedly improved recovery times of animals with metronidazole toxicosis. Diazepam was administered at an initial dose of 0.2–0.5 mg/kg IV followed by 0.3–0.5 mg/kg PO every 8 h for 3 days (Evans et al., 2003). Mean response time (defined as the time to resolution of the debilitating clinical signs) was 4.25 days for untreated dogs to 13.4 h for treated dogs, and mean recovery time (defined as the time to resolution of all residual clinical signs) was 11 days for untreated dogs and 38.8 h for treated dogs (Evans et al., 2003).
Prognosis
Prognosis is generally excellent, with most animals recovering within 2 weeks. However, sometimes recovery can be prolonged in severely affected animals.
Ivermectin and other macrocyclic lactones
Overview
The macrocyclic lactones, including avermectins (abamectin, ivermectin, eprinomectin, doramectin and selamectin) and milbemycins (moxidectin, milbemycin and nemadectin),
L. De Risio
are parasiticides able to kill a wide variety of arthropods and nematodes. Ivermectin is commonly used as heartworm preventative in dogs and cats, as ear miticide in cats, as treatment of sarcoptic and demodectic mange in dogs, and as anthelminthic in ruminants, swine and horses. Intoxication can occur when ivermectin is inadvertently overdosed by the owner or veterinarian or when dogs or cats are exposed to products intended for large animals (including by ingestion of contaminated faeces), which contain a higher concentration of ivermectin than small animal products. In addition, subpopulations of herding type breeds, primarily collies as well as Shetland sheepdogs and Australian shepherds, old English sheepdogs, German shepherds and some mixes of these breeds have a unique sensitivity to macrocyclic lactones and other drugs due to an autosomal recessive mutation in the ATP-binding cassette (ABC) B1 (ABCB1) gene (formerly named multidrug resistance 1 (MDR1) gene). This mutation results in a lack of functional permeability glycoprotein (P-gp) (Nelson et al., 2003; Merola and Eubig, 2012). P-gp is a member of the ATP-binding cassette (ABC) superfamily of transporters and represents an important neuroprotective component of the blood-brain barrier as it limits the entry of macro-cyclic lactones and other xenobiotics into the CNS. The defective P-gp results in accumulation of relatively high concentrations of P-gp-substrate drugs in the CNS, even when relatively low doses of drug are administered.
Mechanism of action
Ivermectin and other macrocyclic lactones cause toxicity in mammals by acting as GABAA-receptor agonists in the CNS. Low CNS concentrations of macrocyclic lactones produce stimulatory CNS effects (e.g. tremors, seizures), whereas higher concentrations result in inhibitory CNS effects (e.g. ataxia, obtundation, stupor, coma).
Clinical presentation
Clinical signs of ivermectin toxicity have been reported at dosages ranging from 0.1 to 0.4 mg/kg PO in ABCB1 gene mutation-susceptible canine breeds, 0.2–2.5 mg/kg PO in non-susceptible canine breeds and of 0.3 mg/kg SC in cats (Merola and Eubig, 2012). Clinical signs include obtundation, disorientation, hypersalivation, muscle tremors, ataxia, blindness, mydriasis, seizures, stupor and coma (Hopper et al., 2002; Nelson et al., 2003; Kenny et al., 2008; Merola et al., 2009). Due to the long half-life of macro-cyclic lactones (from 2 days for ivermectin to 19 days for moxidectin), clinical signs of toxicosis may persist for days to weeks, depending on the agent, dose and the breed involved. Death may occur from respiratory arrest if no therapeutic intervention takes place.
Diagnosis
The clinical diagnosis is based on the history of exposure or over-dosage and clinical signs. A genetic test for the ABCB1-1D mutation is commercially available and can help to identify susceptible dogs. Plasma or stomach contents can be submitted to a veterinary diagnostic laboratory for quantification of ivermectin. Post-mortem diagnosis can be reached by analysis of ivermectin concentration in frozen brain, liver and fat.
Management
There is no specific antidote for ivermectin toxicity. Treatment involves decontamination including multiple doses of activated charcoal (Table 4.1) to interrupt the enterohepatic recirculation of ivermectin; a saline cathartic (Table 4.1), supportive care (intravenous fluids, ventilator support, cardiovascular, respiratory and neurologic monitoring, nursing care), AEMs (see Table 4.1 and Chapters 12 and 24) and myorelaxants in animals with tremors. Induction of emesis (Table 4.1) can be considered in animals with no neurologic signs and recent oral exposure. Historically, GABA agonists AEMs (such as benzodiazepines and barbiturates) were not recommended in animals with ivermectin-induced seizures as ivermectin is also a GABA agonist and exacerbation of clinical signs was a concern. However, this no longer seems to be the case. AEMs with a different mode of action such as levetiracetam (see Table 4.1 and Chapter 16) may be used.
Reactive Seizures
Intravenous lipid emulsion (IVLE) administration has been suggested as a treatment that may shorten the duration of clinical signs of macrocyclic lactones and other lipophilic compound toxicosis. It is hypothesized that the IVLE acts as a ‘lipid sink’ and draws lipophilic compounds into the plasma lipid phase, thereby removing the compound from the target tissues and promoting its elimination (Merola and Eubig, 2012). Intralipid 20% preparation for intravenous infusion administered at 1.5 ml/kg as initial slow bolus followed by a constant rate infusion of 0.25 ml/kg/min over a 30–60 min period has been reported to result in rapid recovery in dogs and one cat with ivermectin toxicosis (Pritchard, 2010; Clarke et al., 2011; Bates et al., 2013; Epstein and Hollingsworth, 2013). IVLE does not seem beneficial to dogs that are homozygus for the ABCB1-1D mutation, possibly because of pre-existent high brain tissue concentration of ivermectin (Wright et al., 2011). Physostigmine and flumazenil may also be beneficial in the treatment of ivermectin toxicity. In severely affected animals intensive nursing care including respiratory, cardiovascular and nutritional support are necessary for several days.
Prognosis
Recovery may occur, however it is often prolonged (>3 weeks). Blind animals may recover vision (Kenny et al., 2008). Exposure to ivermectin doses >5 mg/kg carries a guarded prognosis. Outcome is likely to be more favour-able when decontamination measures are instituted soon after exposure and good supportive care is provided.
Levamisole
Overview
Levamisole is an anthelmintic, microfilaricide and immunostimulant (Bradley, 1976; Vandevelde et al., 1978). Levamisole toxicity has been reported with accidental overdosing (about four times the therapeutic dose) in companion animals.
Mechanism of action
Levamisole is a nicotine-like stimulant producing both muscarinic and nicotinic effects at cholinergic receptors.
Clinical presentation
Clinical signs of levamisole toxicosis include nausea, vomiting, hypersalivation, frequent urination and defecation, muscle tremors, ataxia, anxiety, hyperesthesia with irritability, clonic seizures, obtunded mental status, dyspnoea and cardiac arrhythmias.
Management
Treatment includes gastrointestinal decontamination, AEMs (see Table 4.1 and Chapters 12 and 24) and supportive care (including fluid therapy and oxygenation).
Methylxanthines
Overview
Methylxanthine compounds such as caffeine, theophylline, aminophylline and theobromine are CNS stimulants that are sometimes involved in accidental or malicious poisoning of companion animals (O’Brien, 1998). Intoxication most commonly occurs following ingestion of chocolate, caffeine-based tablets or elixirs. Dark chocolate and cocoa beans have much higher theobromine content than milk or white chocolate. Animals ingesting 20 mg/kg of caffeine or theobromine have mild clinical signs. Ingestion of 60 mg/kg causes seizures.
Mechanism of action
Methylxanthines enhance catecholamine effects both directly and indirectly through phosphodiesterase inhibition, elevation in intracellular cyclic AMP (AMPc) and increased calcium influx into neurons (O’Brien, 1998). This results in increased neuromuscular excitability and a positive inotropic effect. Competitive inhibition of cellular adenosine receptors further increases intracellular AMPc and results in CNS stimulation.
L. De Risio
Clinical presentation
Clinical signs generally occur within 1–2 h post-ingestion and include restlessness, hyperactivity, vomiting, diarrhoea, tachycardia, cardiac arrhythmias, tachypnoea, polydipsia/ polyuria, ataxia, muscle tremors, tonic seizures, hyperthermia, cyanosis, coma and death (Glauberg and Blumenthal, 1983; Tawde et al., 2012).
Diagnosis
Diagnosis is based on the history of methylxanthine (generally theobromine, which is present in chocolate) ingestion, clinical signs and presence of chocolate (or other source of methylxanthine) in gastric content from vomitus or lavage fluids. Methylxanthine concentrations in gastric content, serum, plasma or urine can be analysed.
Management
Treatment is symptomatic and supportive, including induction of emesis (in asymptomatic animals within 2–4 h post-ingestion), activated charcoal with a cathartic (Table 4.1), intravenous fluids to promote methylxanthine excretion, antiarrythmic agents, skeletal muscle relaxants, AEMs (see Table 4.1 and Chapters 12 and 24) and supportive care. Removal of urine though an indwelling catheter prevents reabsorption of the toxin across the bladder mucosa (Hooser and Beasley, 1986).
Prognosis
The prognosis is generally favourable for animals with mild to moderate clinical signs that are treated promptly and aggressively.
Amphetamine and amphetamine-like compounds
Overview
Amphetamine and amphetamine-like compounds such as such as methylphenidate (Ritalin, Concerta), pemoline (Cylert) and dextroamphetamine/amphetamine combinations (Adderall, Dexedrine) are used to treat behaviour disorders in people, including attention deficit disorders in children and adults (Albretsen, 2002). Pets may be exposed by accidental ingestion of their owner’s medications. Toxic doses of amphetamines in dogs and cats can be as low as 1 mg/kg. Pemoline is known to cause clinical signs in dogs at oral doses greater than 2.8 mg/kg (Albretsen, 2002).
Mechanism of action
Amphetamine and amphetamine-like compounds stimulate the release of catecholamine and/or serotonin and prevent their reuptake and metabolism with consequent CNS stimulation (O’Brien, 1998; Stern and Schell, 2012).
Clinical presentation
Clinical signs include hyperactivity, restlessness, irritability or other behavioural changes, mydriasis, muscle tremors, tachycardia or other cardiac arrhythmias, hypertension, tachypnea, hyperthermia, vomiting, diarrhoea and, rarely, seizures (O’Brien, 1998; Albretsen, 2002; Stern and Schell, 2012). Coma and death (attributed to DIC secondary to hyperthermia and respiratory failure) can occur in severe intoxication.
Diagnosis
Diagnosis is based on the history of ingestion, clinical signs, and recovery of pills or capsules in the gastric content from vomitus or lavage fluids. Amphetamines can be detected in urine or plasma by laboratory analysis.
Management
There is no specific antidote for amphetamines or amphetamine-like drug intoxication. Treatment involves decontamination (induction of emesis in asymptomatic animals or gastric lavage followed by the administration of activated charcoal and a cathartic) (Table 4.1), supportive care (including fluid therapy and convective whole body cooling), methocarbamol in animals with tremors and AEMs in seizuring animals (see Table 4.1 and Chapters 12 and 24). Diazepam administration has been reported to increase excitability in some dogs
Reactive Seizures
with amphetamine toxicosis and it is therefore avoided to control hyperactivity, restlessness, irritability or other excitatory behavioural changes; however, it can be used in the acute management of seizures (Albretsen, 2002; Stern and Schell, 2012). Phenothiazine tranquilizers have been recommended to control hyperactivity, restlessness, irritability or other behavioural changes (Stern and Schell, 2012). Acepromazine can initially be administered at 0.05 mg/kg IV and titrated to effect. The dose can be gradually increased to 0.1 to
1.0 mg/kg if clinical signs do not resolve with lower doses. Alternatively, chlorpromazine can initially be administered at 0.5 mg/kg IV and subsequently titrated up to effect. Blood pressure should be monitored at higher doses of phenothiazine to ensure that hypotension does not occur. Animals should be kept in a dark and quiet area and stimulation should be minimized (Stern and Schell, 2012). Treatment of tachycardia or other cardiac arrhythmias may sometimes be required. Urinary acidification has been recommended (to a pH of between 4.5 and 5.5) to enhance the elimination of amphetamines. This can be achieved with ammonium chloride administration at 100 to 200 mg/kg/day PO divided four times daily or ascorbic acid 20–30 mg/kg PO, SQ, IM or IV. Urinary acidification should not be attempted if the animal is acidotic, if acid-base status cannot be monitored regularly, or in case of rhabdomyolosis or acute renal failure (Albretsen, 2002; Stern and Schell, 2012).
Prognosis
Prognosis is generally good with animals receiving prompt and appropriate treatment (Stern and Schell, 2012).
Selective serotonin reuptake inhibitors
Overview
Selective serotonin reuptake inhibitors (SSRIs) such as sertraline (Zoloft), fluoxetine (Prozac), paroxetine (Paxil), and fluvoxamine (Luvox) are often used to treat depression, obsessive-compulsive disorders and other behavioural problems in people (Albretsen, 2002).
Fluoxetine has been used in dogs for the treatment of fear or anxiety, acral lick dermatitis and narcolepsy. Paroxetine has been used to treat urine-spraying problems in cats.
Mechanism of action
As suggested by their name, SSRIs inhibit the reuptake of serotonin at the presynaptic membrane resulting in increased synaptic serotonin levels and overstimulation of serotonin receptors.
Clinical presentation
Clinical signs include obtundation, stupor or coma, anorexia, vomiting, diarrhoea, hyper-salivation, abdominal pain, arrhythmias, hypo- or hypertension, tachypnea, dyspnoea, hyperactivity, hyperaesthesia, tremors, seizures (rarely) and hyperthermia. Signs consistent with CNS depression (e.g. obtundation, stupor or coma) are more common than signs of CNS stimulation (e.g. agitation, aggression, tremors or seizures) (Thomas et al., 2012).
Diagnosis
Diagnosis is based on the history of SSRI ingestion, clinical signs and recovery of pills or capsules in the gastric content from vomitus or lavage fluids.
Management
There are no antidotes available for the treatment of SSRI toxicosis. Treatment is symptomatic and supportive, including induction of emesis (in asymptomatic animals), gastric lavage, activated charcoal with a cathartic (in animals without diarrhoea) (Table 4.1), antiarrythmic agents, skeletal muscle relaxants, AEMs (see Table 4.1 and Chapters 12 and 24) and supportive care. In animals with severe clinical signs, serotonin receptor antagonists such as cyproheptadine at 1.1 mg/ kg PO in dogs and 2.4 mg per cat PO can be administered. The medication can be crushed, mixed with saline and administered rectally to animals unable to take oral medications (Albretsen, 2002).
L. De Risio
Prognosis
The overall prognosis for animals with SSRI toxicosis is excellent with prompt and appropriate treatment (Thomas et al., 2012).
5-Hydroxytryptophan
Overview
5-Hydroxytryptophan (5-HTP), sometimes called Griffonia seed extract, is sold as an over-the-counter dietary supplement. It has been used to help with depression, chronic headaches, obesity and insomnia in people.
Mechanism of action
5-HTP is rapidly converted to serotonin within target cells of the CNS and cardiovascular system, resulting in overstimulation of serotonin receptors.
Clinical presentation
Clinical signs occur from 10 min to 4 h after ingestion and include vomiting, diarrhoea, abdominal pain, hypersalivation, mydriasis, seizures, hyperthermia, obtunded mental status, tremors, hyperesthesia, ataxia, tachycardia and tachypnea (Gwaltney-Brant et al., 2000). In a study including 21 dogs with evidence of accidental 5-HTP ingestion, the minimum toxic dose reported was 23.6 mg/kg and the minimum lethal dose was 128 mg/kg (Gwaltney-Brant et al., 2000).
Diagnosis
Diagnosis is based on history of exposure and clinical signs.
Management
Treatment includes decontamination (by induction of emesis and/or gastric lavage, administration of activated charcoal), AEMs (see Table 4.1 and Chapters 12 and 24) and supportive care including intravenous fluid therapy and convective whole body cooling.
Cyproheptadine, a serotonin antagonist, has been used at 1.1 mg/kg, PO or rectally, every 1 to 4 h until signs resolve. Rectal administration may be preferred when severe vomiting or recent administration of activated charcoal make the oral route unfeasible (Gwaltney-Brant et al., 2000).
Prognosis
The majority of dogs that receive prompt and aggressive treatment recovered within 36 h after exposure (Gwaltney-Brant et al., 2000).
Guarana (Paullinia cupana) and ma huang (Ephedra sinica)
Overview
Guarana (Paullinia cupana), a natural source of caffeine, and ma huang (Ephedra sinica), a natural source of ephedrine, are the main components of an herbal medicine marketed as a weight loss and energy supplement.
Mechanism of action
Caffeine is a methylxanthine and exerts its effect through several mechanisms. It enhances catecholamines release, antagonizes cellular adenosine receptors and inhibits cellular phosphodiesterases thereby increasing intracellular cyclic AMP (AMPc) and calcium influx. Ephedrine is a sympathomimetic alkaloid with a-, b1- and b2-adrenergic agonist activity and CNS stimulatory effect.
Clinical presentation
Accidental ingestion of this herbal supplement has been reported to result in vomiting, hyperactivity, tachycardia, hyperthermia, mydriasis, muscle tremors, behaviour changes and seizures in dogs (Ooms et al., 2001).
Diagnosis
The clinical diagnosis can be supported by detection of caffeine or ephedrine alkaloid in the urine.
Reactive Seizures
Management
Treatment includes decontamination, AEMs (see Table 4.1 and Chapters 12 and 24) and supportive care (including fluid therapy to enhance diuresis and toxin excretion). The use of benzodiazepines is not recommended in pseudoephedrine toxicosis as it has been reported to cause exacerbation of clinical signs.
Prognosis
Most dogs recover with prompt treatment; however, this intoxication can be fatal (Ooms et al., 2001).
References
Albretsen, J.C. (2002) Oral medications. The Veterinary Clinics of North America Small Animal Practice 32, 421–442.
Barker, A.K., Stahl, C., Ensley, S.M. and Jeffery, N.D. (2013) Tremorgenic mycotoxicosis in dogs. Compendium on Continuing Education for Veterinarians 35(2), E1–6.
Barr, A.C. (2006) Household and garden plants. In: Peterson, P.E. and Talcott, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 345–405.
Bath, M.L. (1978) Hexachlorophene toxicity in dogs. Journal of Small Animal Practice 19, 241–244.
Bateman, S.W. and Parent, J.M. (1999) Clinical findings, treatment, and outcome of dogs with status epilepticus or cluster seizures: 156 cases (1990–1995). Journal of American Veterinary Medical Association 215, 1463–1468.
Bates, N., Chatterton, J., Robbins, C., Wells, K., Hughes, J., Stone, M. and Campbell, A. (2013) Lipid infusion in the management of poisoning: a report of 6 canine cases. The Veterinary Record 30, 172(13), 339.
Benitah, N. (2010) Electrolyte disorders: sodium (hyper/hyponatremia). In: Ettinger, S.J. and Feldman, E.C. (eds) Textbook of Veterinary Internal Medicine, 7th edn. Saunders Elsevier, pp. 299–303.
Berent, A.C. and Tobias, K.M. (2009) Portosystemic vascular anomalies. The Veterinary Clinics of North America Small Animal Practice 39(3), 513–541.
Berny, P., Caloni, F., Croubels, S., Sachana, M., Vandenbroucke, V., Davanzo, F. and Guitart, R. (2010) Animal poisoning in Europe. Part 2: Companion animals. Veterinary Journal 183(3), 255–259.
Bismuth, M., Funakoshi, N., Cadranel, J.F. and Blanc, P. (2011) Hepatic encephalopathy: from pathophysiology to therapeutic management. European Journal of Gastroenterology and Hepatology 23(1), 8–22.
Blodgett, D.J. (2006) Organophosphate and Carbamate insecticides. In: Peterson, P.E. and Talcot, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 334–955.
Bradley, R.E. (1976) Levamisole resinate as a Dirofilaria immitis microfilaricide in dogs. Journal of American Veterinary Medical Association 169, 311–316.
Bratton, G.R. and Kowalczyk, D.F. (1989) Lead poisoning. In: Kirk, R.W., Current Veterinary Therapy X: small animal practice. W.B. Saunders, Philadelphia.
Brauer, C., Jambroszy, M. and Tipold, A. (2011) Metabolic and toxic causes of canine seizure disorders: A retrospective study of 96 cases. The Veterinary Journal 187, 272–275.
Caylor, K.B. and Cassimatis, M.K. (2001) Metronidazole neurotoxicosis in two cats. Journal of American Animal Hospital Association 37(3), 258–262.
Christiansen, J.S., Hottinger, H.A., Allen, L., Philipps, L. and Aronson, L.R. (1995) Hepatic microvascular dysplasia in dogs: a retrospective study of 24 cases (1987–1995). Journal of American Veterinary Medical Association 207, 1048–1054.
Clarke, D.L., Lee, J.A., Murphy, L.A. and Reineke, E.L. (2011) Use of intravenous lipid emulsion to treat ivermectin toxicosis in a Border Collie. Journal of American Veterinary Medical Association 15, 239(10), 1328–1333.
Clemmons, R.M. (1990) How do I treat? Acute and subacute organophosphate intoxication in the dog and cat. Progress in Veterinary Neurology 1, 102–103.
Clemmons, R.M., Meyer, D.J. and Sundlof, S.F. (1984) Correction of organophosphate-induced neuromuscular blockade by diphenhydramine. American Journal Veterinary Research 45(10), 2167–2169.
Connally, H.E., Thrall, M.A., Forney, S.D., Grauer, G.F. and Hamar, D.W. (1996) Safety and efficacy of 4-methylpyrazole for treatment of suspected or confirmed ethylene glycol intoxication in dogs: 107 cases (1983-1995). Journal of American Veterinary Medical Association 209, 1880–1883.
L. De Risio
D’Anjou, M.A., Penninck, D., Cornejo, L. and Pibarot, P. (2004) Ultrasonographic diagnosis of portosystemic shunting in dogs and cats. Veterinary Radiology and Ultrasound 45(5), 424–437.
DeClementi, C. and Sobczak, B.R. (2012) Common rodenticide toxicoses in small animals. Veterinary Clinics of North America Small Animal Practice 42(2), 349–360.
de Morais, H.A. and DiBartola, S.P. (2008a) Hyponatremia: a quick reference. The Veterinary Clinics of North America Small Animal Practice 38(3), 491–495.
de Morais, H.A. and DiBartola, S.P. (2008b) Hypernatremia: a quick reference. The Veterinary Clinics of North America Small Animal Practice 38(3), 485–489.
Dial, S.M., Thrall, M.A. and Hamar, D.W. (1994a) Efficacy of 4-methylpyrazole for treatment of ethylene glycol intoxication in dogs. American Journal of Veterinary Research 55, 1762–1770.
Dial, S.M., Thrall, M.A. and Hamar, D.W. (1994b) Comparison of ethanol and 4-methylpyrazole as treatments for ethylene glycol intoxication in cats. American Journal of Veterinary Research 55, 1771–1782. Dorman, D.C. and Fikes, J.D. (1993) Diagnosis and therapy of neurotoxicological syndromes in dogs and cats: selected syndromes induced by pesticides, part 2. Progress in Veterinary Neurology 4(4), 111–120.
Dorman, D.C., Parker, A.J. and Buck, W.B. (1990a) Bromethalin toxicosis in the dog. Part I: clinical effects. Journal of American Animal Hospital Association 26, 589.
Dorman, D., Parker, A. and Buck, W. (1990b) Bromethalin toxicosis in the dog. Part II: Selected treatments for the toxic syndrome. Journal of American Animal Hospital Association 26, 595–598.
Dorman, D.C., Parker, A.J. and Dye, J.A. (1990c) Bromethalin neurotoxicosis in the cat. Progress in Veterinary Neurology 1, 189.
Dow, S.W., LeCouteur, R.A., Poss, M.L. and Beadleston, D. (1989) Central nervous system toxicosis associated with metronidazole treatment of dogs: five cases (1984-1987). Journal of American Veterinary Medical Association 195(3), 365–368.
Drobatz, K.J. and Casey, K.K. (2000) Eclampsia in dogs: 31 cases (1995–1998). Journal of the American Veterinary Medical Association 217, 216–219.
Edwards, C., Beattie, K.A., Scrimgeour, C.M. and Codd, G.A. (1992) Identification of anatoxin-A in benthic cyanobacteria (blue-green algae) and in associated dog poisonings at Loch Insh, Scotland. Toxicon: Official Journal of the International society of Toxinology 30(10), 1165–1175.
Elford, J., Monteiro, R., McKee, M. and Behr, S. (2012) Possible case of blue-green algae (Cyanobacteria) toxicity. The Veterinary Record 24, 171(21), 541–542.
Ellenhorn, M.J., Schonwald, S. and Ordog, G. (1997) Ellenhorn’s Medical Toxicology: diagnosis and treatment of human poisoning, 2nd edn. Williams & Wilkins, Baltimore, Maryland.
Epstein, S.E. and Hollingsworth, S.R. (2013) Ivermectin-induced blindness treated with intravenous lipid therapy in a dog. Journal of Veterinary Emergency and Critical Care 23(1), 58–62.
Eriksen, G.S., Jäderlund, K.H., Moldes-Anaya, A., Schönheit, J., Bernhoft, A., Jaeger, G., Rundberget, T. and Skaar,
I. (2010) Poisoning of dogs with tremorgenic Penicillium toxins. Medical Mycology 48(1), 188–196.
Evans, J., Levesque, D., Knowles, K., Longshore, R. and Plummer, S. (2003) Diazepam as a Treatment for Metronidazole Toxicosis in Dogs: A Retrospective Study of 21 Cases. Journal of Veterinary Internal Medicine 17, 304–310.
Faassen, E.J., Harkema, L., Begeman, L. and Lurling, M. (2012) First report of (homo)anatoxin-a and dog neurotoxicosis after ingestion of benthic cyanobacteria in The Netherlands. Toxicon: Official Journal of the International Society of Toxicology 1, 60(3), 378–384.
Fenner, W.R. (1995) Uremic encephalopathy. In: Bonagura, D.J. (ed.) Kirk’s Current Veterinary Therapy XII (Small Animal Practice). W.B. Saunders, Philadelphia, Pennsylvania, pp. 1158–1161.
Fernandez, A.L., Lee, J.A., Rahilly, L., Hovda, L., Brutlag, A.G. and Engebretsen, K. (2011) The use of intravenous lipid emulsion as an antidote in veterinary toxicology. Journal of Veterinary Emergency and Critical Care 21(4), 309–320.
Frankel, D., Seim, H., MacPhail, C. and Monnet, E. (2006) Evaluation of cellophane banding with and without intraoperative attenuation for treatment of congenital extrahepatic portosystemic shunts in dogs. Journal of American Veterinary Medical Association 228, 1355–1360.
Fryer, K.J., Levine, J.M., Peycke, L.E., Thompson, J.A. and Cohen, N.D. (2011) Incidence of Postoperative Seizures with and without Levetiracetam Pretreatment in Dogs Undergoing Portosystemic Shunt Attenuation. Journal of Veterinary Internal Medicine 25, 1379–1384.
Garosi, L.S., Dennis, R. and Platt, S.R. (2003) Thiamine deficiency in a dog: clinical, clinicopathologic and magnetic resonance imaging findings. Journal of Veterinary Internal Medicine 17, 719–723. Garza, A., Vega, R. and Soto, E. (2006) Cellular mechanisms of lead neurotoxicity. Medical Science Monitor: International Medical Journal of Experimental and Clinical Research 12(3), RA57–65.
Glauberg, A. and Blumenthal, H.P. (1983) Chocolate poisoning in the dog. Journal of American Animal Hospital Association 19, 246–248.
Goh, C.S., Hodgson, D.R., Fearnside, S.M., Heller, J. and Malikides, N. (2005) Sodium monofluoroacetate (Compound 1080) poisoning in dogs. Australian Veterinary Journal 83(8), 474–479.
Goutal, C.M., Brugmann, B.L. and Ryan, K.A. (2012) Insulinoma in dogs: a review. Journal of American Animal Hospital Association 48(3), 151–163.
Gray, S.L., Lee, J.A., Hovda, L.R. and Brutlag, A.G. (2011) Potential zinc phosphide rodenticide toxicosis in dogs: 362 cases (2004–2009). Journal of American Veterinary Medical Association 239, 646–651.
Gugger, M., Lenoir, S., Berger, C., Ledreux, A., Druart, J.C., Humbert, J.F., Guette, C. and Bernard, C. (2005) First report in a river in France of the benthic cyanobacterium Phormidium favosum producing anatoxina associated with dog neurotoxicosis. Toxicon: Official Journal of the International Society of Toxinology 1, 45(7), 919–928.
Gwaltney-Brant, S. and Meadows, I. (2012) Use of intravenous lipid emulsions for treating certain poisoning cases in small animals. Veterinary Clinics of North America Small Animal Practice 42(2), 251–262.
Gwaltney-Brant, S.M., Albretsen, J.C. and Khan, S.A. (2000) 5-Hydroxytryptophan toxicosis in dogs: 21 cases (1989-1999). Journal of American Veterinary Medical Association 216, 1937–1940.
Hansen, S.R. (2006) Pyrethrins and pyrethroids. In: Peterson, P.E. and Talcott, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 1002–1010.
Hardy, R.M. (1990) Pathophysiology of hepatic encephalopathy. Seminars in Veterinary Medicine and Surgery 5, 100–106.
Hatch, R. (1988) Antinematodal drugs. In: Booth, N. and McDonald, L. (eds) Veterinary Pharmacology and Therapeutics. Iowa State University Press, Ames, Iowa, pp. 882–927.
Haworth, M.D. and Smart, L. (2012) Use of intravenous lipid therapy in three cases of feline permethrin toxicosis. Journal of Veterinary Emergency and Critical Care 22(6), 697–702.
Hess, R.S. (2010) Insulin-secreting islet cell neoplasia. In: Disorders of the Parothyd glands. In: Ettinger, S.J. and Feldman, E.C. (eds) Textbook of Veterinary Internal Medicine, 7th edn. Saunders Elsevier, pp. 1779–1782.
Hooser, S.B. and Beasley, V.R. (1986) Methylxanthine poisoning (chocholate and caffeine toxicosis). In: Kirk, R.W. (ed.) Current Veterinary Therapy IX. W.B. Saunders, Philadelphia, pp. 191–192.
Hooser, S.B. and Talcott, P.A. (2006) Cyanobacteria. In: Peterson, P.E. and Talcott, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 685–689.
Hopper, K., Aldrich, J. and Haskins, S.C. (2002) Ivermectin toxicity in 17 collies. Journal of Veterinary Internal Medicine 16, 89–94.
Jull, P., De Risio, L., Horton, C. and Volk, H.A. (2011) Effect of prolonged status epilepticus as a result of intoxication on epileptogenesis in a UK canine population. Veterinary Record 169, 361.
Kaplan, A. and Whelan, M. (2012) The use of IV lipid emulsion for lipophilic drug toxicities. Journal of American Animal Hospital Association 48(4), 221–227.
Kenny, P.J., Vernau, K.M., Puschner, B. and Maggs, D.J. (2008) Retinopathy associated with ivermectin toxicosis in two dogs. Journal of American Veterinary Medical Association 233, 279–284.
Khan, S.A., Schell, M.M. and Trammel, H.L. (1999) Ethylene glycol exposures managed by the ASPCA National Animal Poison Control Center from July 1995 to December 1997. Veterinary and Human Toxicology 41, 403–406.
Kilpatrick, S., Gow, A.G., Foale, R.D., Tappin, S.W., Carruthers, H., Reed, N., Yool, D.A., Woods, S., Marques, A.I., Jalan, R., Mellanby, R.J. (2014) Plasma cytokine concentrations in dogs with a congenital portosystemic shunt. Veterinary Journal 200(1), 197–199.
Knecht, C.D., Crabtree, J. and Katherman, A. (1979) Clinical, clinicopathologic, and electroencephalographic features of lead poisoning in dogs. Journal of American Veterinary Medical Association 175, 196–201.
Knight, T.E. and Kumar, M.S. (2003) Lead toxicosis in cats-a review. Journal of Feline Medicine and Surgery 5(5), 249–255.
Knight, T.E., Kent, M. and Junk, J.E. (2001) Succimer for treatment of lead toxicosis in two cats. Journal of American Veterinary Medical Association 218, 1946–1948.
Kuo, K., Odunayo, A. (2013) Adjunctive therapy with intravenous lipid emulsion and methocarbamol for permethrin toxicity in 2 cats. Journal of Veterinary Emergency and Critical Care 23(4), 436–441.
Kyles, A.E., Gregory, C.R., Wooldridge, J.D., Mathews, K.G., Aronson, L.R., Bernsteen, L. and Ilkiw, J.E. (1999) Management of hypertension controls postoperative neurologic disorders after renal transplantation in cats. Veterinary Surgery 28(6), 436–441.
L. De Risio
Kyles, A.E., Hardie, E.M., Mehl, M. and Gregory, C.R. (2002) Evaluation of ameroid ring constrictors for the management of single extrahepatic portosystemic shunts in cats: 23 cases (1996–2001). Journal of American Veterinary Medical Association 220, 1341–1347.
Lee, K.C.L., Lipscomb, V.J., Lamb, C.R., Gregory, S.P., Guitian, J. and Brockman, D.J. (2006) Association of portovenographic findings with outcome in dogs receiving surgical treatment for single congenital portosystemic shunts: 45 cases (2000–2004). Journal of American Veterinary Medical Association 229, 1122–1129.
Leifer, C.E., Peterson, M.E. and Matus, R.E. (1986) Insulin-secreting tumor: diagnosis and medical and surgical management in 55 dogs. Journal of American Veterinary Medical Association 188(1), 60–64.
Lidsky, T.I. and Schneider, J.S. (2003) Lead neurotoxicity in children: basic mechanisms and clinical correlates. Brain 126(Pt 1), 5–19.
Lipscomb, V.L., Jones, H.J. and Brockman, D.J. (2007) Complications and long term outcomes of the ligation of congenital portosystemic shunts in 49 cats. Veterinary Record 160, 465–470.
Lowes, N.R., Smith, R.A. and Beck, B.E. (1992) Roquefortine in the stomach contents of dogs suspected of strychnine poisoning in Alberta. Canine Veterinary Journal 33, 535–538.
Maddison, J.E. (1992) Hepatic encephalopathy: current concepts of the pathogenesis. Journal of Veterinary Internal Medicine 6, 341–353.
Mai, W. and Weisse, C. (2011) Contrast-enhanced portal magnetic resonance angiography in dogs with suspected congenital portal vascular anomalies. Veterinary Radiology and Ultrasound 52(3), 284–288.
Markovich, J.E., Freeman, L.M., Heinze, C.R. (2014) Analysis of thiamine concentrations in commercial canned foods formulated for cats. Journal of American Animal Hospital Association 244(2), 175–179.
Marks, S.L., Lipsitz, D., Vernau, K.M., Dickinson, P.J., Draper, W., Larsen, J.A. and Fascetti, A.J. (2011) Reversible encephalopathy secondary to thiamine deficiency in 3 cats ingesting commercial diets. Journal of Veterinary Internal Medicine 25(4), 949–953.
Merola, V., Khan, S. and Gwaltney-Brant, S. (2009) Ivermectin toxicosis in dogs: A retrospective study. Journal of American Animal Hospital Association 45, 106–111.
Merola, V.M. and Eubig, P.A. (2012) Toxicology of avermectins and milbemycins (macrocylic lactones) and the role of P-glycoprotein in dogs and cats. Veterinary Clinics of North America Small Animal Practice 42(2), 313–333.
Moon, S.J., Kim, J.W., Kang, B.T., Lim, C.Y. and Park, H.M. (2012) Magnetic resonance imaging findings of hepatic encephalopathy in a dog with a portosystemic shunt. Journal of Veterinary Medicine and Science 74(3), 361–366.
Morgan, R.V. (1994) Lead poisoning in small companion animals: an update (1987-1992). Veterinary Human Toxicology 36, 18–22.
Murphy, M.J. (2002) Rodenticides. The Veterinary Clinics of North America Small Animal Practice 32(2), 469–484.
Nelson, N.C. and Nelson, L.L. (2011) Anatomy of extrahepatic portosystemic shunts in dogs as determined by computed tomography angiography. Veterinary Radiology and Ultrasound 52(5), 498–506.
Nelson, O.L., Carsten, E., Bentjen, S.A. and Mealey, K.L. (2003) Ivermectin toxicity in an Australian shepherd dog with MDR1 mutation associated with ivermectin sensitivity in collies. Journal of Veterinary Internal Medicine 17, 354–356.
O’Brien, D.P. (1998) Toxic and metabolic causes of seizures. Clinical Techniques in Small Animal Practice 13, 159–166.
O’Hagan, B.J. (2004) Fluoroacetate poisoning in seven domestic dogs. Australian Veterinary Journal 82(12), 756–758.
Ooms, T.G., Khan, S.A. and Means, C. (2001) Suspected caffeine and ephedrine toxicosis resulting from ingestion of an herbal supplement containing guarana and ma huang in dogs: 47 cases (1997–1999). Journal of American Veterinary Medical Association 218(2), 225–229.
Pákozdy, A., Leschnik, M., Tichy, A.G. and Thalhammer, J.G. (2008) Retrospective clinical comparison of idiopathic versus symptomatic epilepsy in 240 dogs with seizures. Acta Veterinaria Hungarica 56(4), 471–483.
Pákozdy, A., Leschnik, M., Sarchahi, A.A., Tichy, A.G. and Thalhammer, J.G. (2010) Clinical comparison of primary versus secondary epilepsy in 125 cats. Journal of Feline Medicine and Surgery 12, 910–916.
Palus, V., Penderis, J., Jakovljevic, S. and Cherubini, G.B. (2010) Thiamine deficiency in a cat: resolution of MRI abnormalities following thiamine supplementation. Journal of Feline Medicine and Surgery 12(10), 807–810.
Parton, K. (2006) Sodium monofluoracetate (1080). In: Peterson, P.E. and Talcott, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 1055–1062.
Penderis, J., McConnell, J.F. and Calvin, J. (2007) Magnetic resonance imaging features of thiamine deficiency in a cat. Veterinary Record 160, 270–272.
Peterson, M.E. and Roberts, B.K. (2006) In: Peterson, P.E. and Talcott, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 345–405.
Platt, S.R. and Haag, M. (2002) Canine status epilepticus: A retrospective study of 50 cases. Journal of Small Animal Practice 43, 151–153.
Podell, M., Fenner, W.R. and Powers, J.D. (1995) Seizure classification in dogs from a nonreferral-based population. Journal of the American Veterinary Medical Association 206, 1721–1728.
Poh, Z. and Chang, P.E. (2012) A current review of the diagnostic and treatment strategies of hepatic encephalopathy. International Journal of Hepatology 2012:480309. doi: 10.1155/2012/480309. Epub 2012 Oct 21.
Pritchard, J. (2010) Treating ivermectin toxicity in cats. Veterinary Record 12, 166(24), 766.
Proudfoot, A.T. (2009) Aluminum and zinc phosphide poisoning. Clinical Toxicology 47, 89–100.
Proudfoot, A.T., Bradberry, S.M. and Vale, J.A. (2006) Sodium fluoroacetate poisoning. Toxicology Reviews 25(4), 213–219.
Puschner, B. (2006) Metaldehyde. In: Peterson, P.E. and Talcott, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 830–839.
Puschner, B., Pratt, C. and Tor, E.R. (2010) Treatment and diagnosis of a dog with fulminant neurological deterioration due to anatoxin–a intoxication. Journal of Veterinary Emergency and Critical Care 20(5), 518–522.
Raisbeck, M.F. (2006) Organochlorine Pesticides. In: Peterson, P.E. and Talcott, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 934–940.
Ramsey, D.T., Casteel, S.W., Faggella, A.M., Chastain, C.B., Nunn, J.W. and Schaeffer, D.J. (1996) Use of orally administered succimer (meso-2,3- imercaptosuccinic acid) for treatment of lead poisoning in dogs. Journal of American Veterinary Medical Association 208, 371–375.
Reeves, M.P. (2004) A retrospective report of 90 dogs with suspected cane toad (Bufo marinus) toxicity. Australian Veterinary Journal 82(10), 608–611.
Roberts, B.K., Aronsohn, M.G., Moses, B.L., Burk, R.L., Toll, J. and Weeren, F.R. (2000) Bufo marinus intoxication in dogs: 94 cases (1997–1998). Journal of American Veterinary Medical Association 216, 1941–1944.
Rosendale, M.E. (2002) Decontamination strategies. Veterinary Clinics of North America Small Animal Practice 32, 311–321.
Rothuizen, J. (2009) Important Clinical Syndromes Associated with Liver Disease. Veterinary Clinics of North America Small Animal Practice 39, 419–437.
Ruland, K., Fischer, A. and Hartmann, K. (2010) Sensitivity and specificity of fasting ammonia and serum bile acids in the diagnosis of portosystemic shunts in dogs and cats. Veterinary Clinical Pathology 39(1), 57–64.
Rusbridge, C. (2005) Diagnosis and control of epilepsy in the cat. In Practice 27, 208–214.
Schenck, P.A. and Chew, D.J. (2008) Hypocalcemia. Veterinary Clinics of North America Small Animal Practice 38(3), 455–458.
Scherk, J.R., Brainard, B.M., Collicutt, N.B., Bush, S.E., Almy, F.S. and Koenig, A. (2013) Preliminary evaluation of a quantitative ethylene glycol test in dogs and cats. Journal of Veterinary Diagnostic Investigations 25(2), 219–225.
Schriefl, S., Steinberg, T.A., Matiasek, K., Ossig, A., Fenske, N. and Fischer, A. (2008) Etiologic classification of seizures, signalment, clinical signs, and outcome in cats with seizure disorders: 91 cases (2000-2004). Journal of the American Veterinary Medical Association 233, 1591–1597.
Smith, R.A. and Lang, D.G. (2000) Rapid determination of ethylene glycol and glycolic acid in biological fluids. Veterinary and Human Toxicology 42, 358–360.
Steinmetz, S., Tipold, A., Löscher, W. (2013) Epilepsy after head injury in dogs: a natural model of posttraumatic epilepsy. Epilepsia 54(4), 580–588.
Stern, L.A. and Schell, M. (2012) Management of attention-deficit disorder and attention-deficit/hyperactivity disorder drug intoxication in dogs and cats. Veterinary Clinics of North America Small Animal Practice 42(2), 279–287.
Sura, P.A., Tobias, K.M., Morandi, F., Daniel, G.B. and Echandi, R.L. (2007) Comparison of 99mTcO4(-) trans-splenic portal scintigraphy with per-rectal portal scintigraphy for diagnosis of portosystemic shunts in dogs. Veterinary Surgery 36(7), 654–660.
Tawde, S.N., Puschner, B., Albin, T., Stump, S. and Poppenga, R.H. (2012) Death by caffeine: presumptive malicious poisoning of a dog by incorporation in ground meat. Journal of Medical Toxicology: Official Journal of American College of Medical Toxicology 8(4), 436–440.
Thomas, D.E., Lee, J.A. and Hovda, L.R. (2012) Retrospective evaluation of toxicosis from selective serotonin reuptake inhibitor antidepressants: 313 dogs (2005-2010). Journal of Veterinary Emergency and Critical Care 22(6), 674–681.
Thompson, J.P., Senior, D.F. and Pinson, D.M. (1987) Neurotoxicosis associated with the use of hexachlorophene in a cat. Journal of American Veterinary Medical Association 190, 1311–1312.
Thrall, M.A., Connally, H.E., Grauer, G.F. and Hamar, D. (2006) Ethylene glycol. In: Peterson, P.E., Talcott, P.A. (eds) Small Animal Toxicology, 2nd edn. Elsevier Saunders, St Louis, Missouri, pp. 702–726.
Tisdall, P., Hunt, G.B., Youmans, K.R. and Malik, R. (2000) Neurological dysfunction in dogs following attenuation of congenital extrahepatic portosystemic shunts. Journal of Small Animal Practice 41, 539–546.
Torisu, S., Washizu, M., Hasegawa, D. and Orima, H. (2005) Brain magnetic resonance imaging characteristics in dogs and cats with congenital portosystemic shunts. Veterinary Radiology and Ultrasound 46(6), 447–451.
Van Hee, P., Neels, H., De Doncker, M., Vrydags, N., Schatteman, K., Uyttenbroeck, W., Hamers, N., Himpe D. and Lambert, W. (2004) Analysis of gamma-hydroxybutyric acid, DL-lactic acid, glycolic acid, ethylene glycol and other glycols in body fluids by a direct injection gas chromatography-mass spectrometry assay for wide use. Clinical Chemistry and Laboratory Medicine 42(11), 1341–1345.
Vandevelde, M., Boring, J.G., Hoff, E.J. and Gingerich, D.A. (1978) The effect of levamisole on the canine central nervous system. Journal of Neuropathology and Experimental Neurology 37, 165–173.
Ward, B., Jones, B. and Rubin, G. (1973) Hexachlorophene toxicity in dogs. Journal of American Animal Hospital Association 9, 167.
Wismer, T. and Means, C. (2012) Toxicology of newer insecticides in small animals. Veterinary Clinics of North America Small Animal Practice 42(2), 335–347.
Wolf, A.M. (1980) Canine uremic encephalopathy. Journal of American Animal Hospital Association 16, 735–738.
Wright, H.M., Chen, A.V., Talcott, P.A., Poppenga, R.H. and Mealey, K.L. (2011) Intravenous fat emulsion as treatment for ivermectin toxicosis in three dogs homozygous for the ABCB1-1b gene mutation. Journal of Veterinary Emergency and Critical Care 21(6), 666–672.
Yas-Natan, E., Segev, G. and Aroch, I. (2007) Clinical, neurological and clinicopathological signs, treatment and outcome of metaldehyde intoxication in 18 dogs. Journal of Small Animal Practice 48(8), 438–443.
Young, K.L., Villar, D., Carson, T.L., Ierman, P.M., Moore, R.A. and Bottoff, M.R. (2003) Tremorgenic mycotoxin intoxication with penitrem A and roquefortine in two dogs. Journal of American Veterinary Medical Association 222(1), 52–53, 35.
Zimmermann, R., Hülsmeyer, V., Sauter-Louis, C. and Fischer, A. (2009) Status epilepticus and epileptic seizures in dogs. Journal of Veterinary Internal Medicine 23(5), 970–976.
Zook, B.C., Carpenter, J.L. and Leeds, E.B. (1967) Lead poisoning in dogs. Journal of American Veterinary Medical Association 155, 1329–1342.
5 Structural Epilepsy
Luisa De Risio
Neurology/ Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Structural epilepsy is caused by a known and identifiable structural forebrain disorder such as vascular, inflammatory/infectious, traumatic, anomalous/developmental, neoplastic and degenerative diseases. Reported prevalence of structural epilepsy in dogs and cats varies among studies ranging from 25–38% in dogs and 34–87% in cats (Quesnel et al., 1997; Bateman and Parent, 1999; Platt and Haag, 2002; Pákozdy et al., 2008; Schriefl et al., 2008; Zimmermann et al., 2009; Steinmetz et al., 2013). Dogs and cats with structural epilepsy usually present with neurological signs (other than seizures) interictally. However, focal lesions in particular areas of the brain (‘clinically silent regions’), such as olfactory bulb, frontal, and pyriform lobes can result in seizure activity without any other neurological signs. In addition, seizures may be the first clinical sign or the only abnormality the pet owner recognizes, at least initially. The signalment, history, disease onset and course and neuroanatomic localization can help to formulate the most appropriate differential diagnosis list. Diagnostic investigations (see Chapter 10) are aimed at identifying the underlying aetiology of the seizures and, if present, of the other neurological signs. Treatment is aimed at the underlying aetiology of the structural brain disease and seizure control with antiepileptic medications (AEMs). Commonly used first-line AEMs are phenobarbital (PB) and potassium bromide (KBr) (see Chapters 12, 13 and 14). Loading or combination with other AEMs with shorter half-lives (such as levetiracetam or zonisamide, see Chapters 15 and 16) may be required to obtain timely clinical efficacy. New generation AEMs may also be used as first-line monotherapy when PB- or Br-related excessive sedation is a concern (e.g. dogs with brain tumours). The choice and dose of the most appropriate AEM is influenced not only by the underlying disease but also by its possible systemic manifestations or concurrent disorders (e.g. PB is contraindicated in animals with hepatic dysfunction; dose reduction of renally excreted AEMs is necessary in animals with impaired renal function). In addition, interactions between AEMs and treatment of the underlying cause of structural epilepsy have to be considered
(e.g. in dogs, PB can alter the pharmacokinetics and as a consequence may decrease therapeutic effect of corticosteroids, cyclosporine, metronidazole and other medications) (see Chapter 13). Animals with structural epilepsy may present with cluster seizures or status epilepticus (Bateman and Parent, 1999; Zimmermann et al., 2009). Detailed information on pathophysiology and management of cluster seizures and status epilepticus are presented in Chapter 23 and 24, respectively.
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
Disease groups resulting in structural epilepsy are presented in order according to the acronym VITAMIN D.
Vascular
Cerebrovascular accidents (ischaemic, haemorrhagic)
Cerebrovascular accidents (CVAs) result from cerebrovascular disease which involves any pathological process of the blood vessels supplying the brain (Wessmann et al., 2009). CVA, also termed stroke, is characterized by non-progressive (or rapidly progressive) intracranial neurological signs with peracute (6 h) to acute (7–24 h) onset and duration of at least 24 h (Victor and Ropper, 2001). When the neurological signs last less than 24 h, the event is referred to as a transient ischaemic attack (TIA) (Victor and Ropper, 2001). TIAs may precede a CVA.
CVAs can be broadly classified as:
Ischaemic CVA can be classified by the territory that the affected blood vessel supplies, the size of the vessel (e.g. territorial infarct with large arterial vessel disease, Fig. 5.1a–e; and lacunar infarct with small perforating arterial vessel disease, Fig. 5.2a–e), the age of the infarct (e.g. recent, organizing), the presence of secondary haemorrhage, the pathogenesis of the stroke (e.g. thrombotic, embolic, haemodynamic) and the suspected underlying aetiology.
Haemorrhagic CVA can be classified according to the anatomical site of the haemorrhage
(e.g. epidural, subdural, Fig. 5.3a–d; subarachnoid; intraparenchymal, Fig. 5.4a–f; intraventricular), size of the lesion (e.g. small, large) and the age of the lesion or the suspected underlying aetiology (Wessmann et al., 2009). Several disorders can predispose to ischaemic or haemorrhagic CVA (Box 5.1).
The most commonly reported concurrent medical conditions in dogs with ischaemic CVA include hyperadrenocorticism, chronic renal disease, hypothyroidism and hypertension (Garosi et al., 2005a). Reported concurrent medical conditions in dogs with haemorrhagic CVA include Angiostrongylus vasorum infection, primary or secondary brain tumours, hypertension, hyperadrenocorticism, chronic renal disease and hypothyroidism (Lowrie et al., 2012). A concurrent medical condition has been identified in approximately 50% and 44% of dogs with ischaemic and haemorrhagic strokes, respectively (Garosi et al., 2005a; Lowrie et al., 2012).
Reports of ischaemic or haemorrhagic strokes in cats are limited (Cherubini et al., 2007; Altay et al., 2011). Reported concurrent medical conditions include hyperthyroidism, hypertrophic cardiomyopathy, hepatic and renal disease (Altay et al., 2011). The term feline ischemic encephalopathy has been used to describe cases of peracute onset of neurological signs (including seizures) consistent with a unilateral (focal) forebrain lesion caused by ischaemia associated with Cuterebra larval migration through the cerebrum. It has been suggested that the migrating parasite or the host response leads to vasospasm in the cerebral vasculature, typically the middle cerebral artery, resulting in focal cerebral ischaemia (Glass et al., 1998; Williams et al., 1998).
Clinical signs
Neurological signs in animals with CVA have typically a peracute (6 h) to acute (7–24 h) onset and are nonprogressive or rapidly progressive and subsequently regressive in their evolution. Progression of neurological signs for 24 to 72 h can occur due to worsening cerebral oedema and haemorrhage. Once the neurological signs plateau, they gradually improve in most cases but CVA can be fatal. In animals with ischaemic CVA neurological signs commonly refer to a focal and unilateral intracranial anatomic neurolocalization (Garosi, 2010). In animals with haemorrhagic strokes, neurological signs may refer to more than one intracranial neuroanatomic localization (e.g. forebrain, brainstem or cerebellum) as the haemorrhage usually involves the territory of more than one artery and intracranial pressure (ICP) may be increased.
Structural Epilepsy
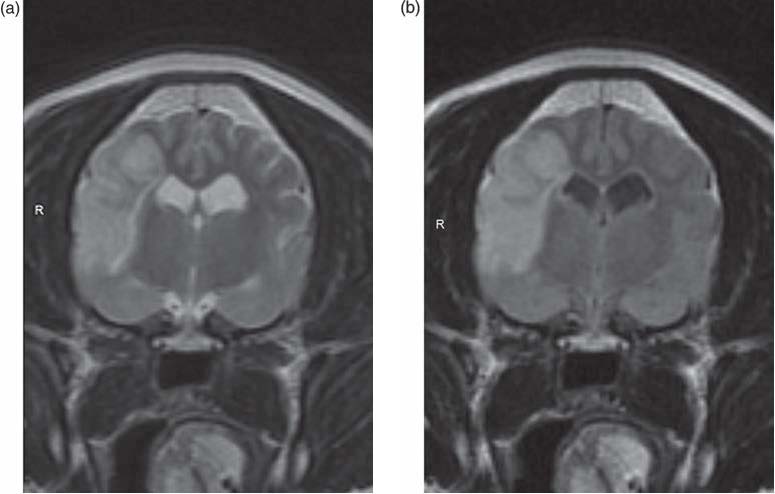
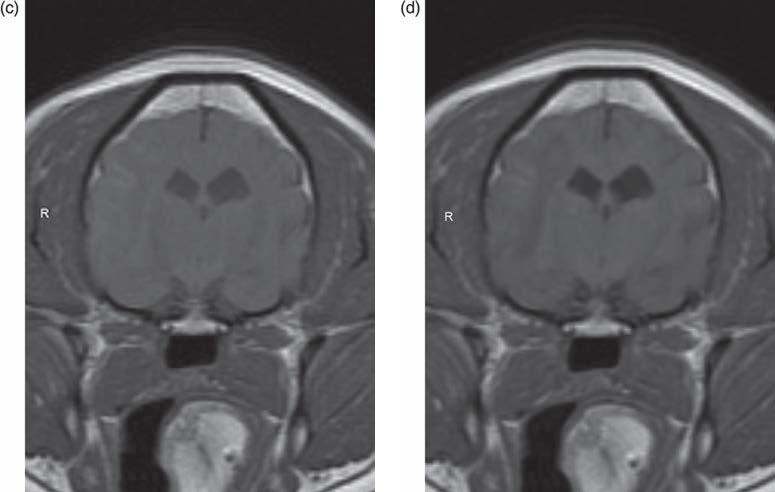
Fig. 5.1. MRI of a 12-year-old, male West Highland white terrier with peracute onset of right-sided forebrain signs (severe obtundation, disorientation and compulsive circling to the right, left-sided hemiparesis, absent postural reactions in the left thoracic and pelvic limbs, decreased menace response on the left eye and decreased facial sensation on the left side of the face). Transverse T2W (a), FLAIR (b), T1W (c), T1WC (d) and dorsal DWI (e) images show a large sharply demarcated T2W, FLAIR and DWI hyperintense, T1W hypointense and non-contrast-enhancing area in the right cerebral cortex involving the parietal lobe and the rostral occipital cortex (e). The MRI features of this lesion are strongly suggestive of a territorial ischaemic infarct affecting the right cerebrum in the territory of the middle cerebral artery.

Fig. 5.1. Continued. Transverse T2W (a), FLAIR (b), T1W (c), T1WC (d) and dorsal DWI (e) images show a large sharply demarcated T2W, FLAIR and DWI hyperintense, T1W hypointense and noncontrast-enhancing area in the right cerebral cortex involving the parietal lobe and the rostral occipital cortex (e). The MRI features of this lesion are strongly suggestive of a territorial ischaemic infarct affecting the right cerebrum in the territory of the middle cerebral artery.
Box 5.1. Disorders predisposing to ischaemic or haemorrhagic CVA.
Ischaemic CVA:
Structural Epilepsy

Fig. 5.2. MRI of a 12-year-old, male neutered, crossbreed with peracute onset of left-sided forebrain signs (severe obtundation, compulsive circling to the left, absent postural reactions in the right thoracic and pelvic limbs, decreased menace response on the right eye and decreased facial sensation on the right side of the face). Transverse T2W (a), FLAIR (b), T1W (c), T1WC (d) and dorsal DWI (e) images show a small relatively sharply demarcated area in the left thalamus, which appears hyperintense on T2W, FLAIR and DWI, iso- to hypo-intense on T1W, and non-contrast enhancing. The MRI features of this lesion are strongly suggestive of a lacunar ischaemic infarct resulting from obstruction of a small perforating artery in the left thalamus.

Fig. 5.2. Continued. Transverse T2W (a), FLAIR (b), T1W (c), T1WC (d) and dorsal DWI (e) images show a small relatively sharply demarcated area in the left thalamus, which appears hyperintense on T2W, FLAIR and DWI, iso- to hypo-intense on T1W, and non-contrast enhancing. The MRI features of this lesion are strongly suggestive of a lacunar ischaemic infarct resulting from obstruction of a small perforating artery in the left thalamus.

Structural Epilepsy


(a) and FLAIR (e and f), and mild peripheral contrast enhancement on T1WC (d). These signal changes were suggestive of intraparenchymal haemorrhage in the right parietal lobe. FLAIR images (e and f) show marked hyperintensity (most likely marked secondary vasogenic oedema) within the corona radiata. The dog had positive faecal culture for Angiostrongylus vasorum.

In animals with CVA affecting the forebrain, seizures can occur immediately or several weeks later and are often recurrent (see section on poststroke seizures and epilepsy) (Garosi et al., 2005a; Lowrie et al., 2012). Ocular fundic examination should be performed in all animals with suspected CVA as it may reveal tortuous retinal vessels (suggestive of systemic hypertension) haemorrhage (suggestive of coagulopathy or systemic hypertension) or papilledema (suggestive of increased ICP) (Garosi, 2010).
Structural Epilepsy
CVA can recur and relapses are most frequent in dogs where an underlying cause is identified but it is difficult to treat (Garosi et al., 2005a).
Post-stroke seizures and epilepsy
Seizures can occur secondary to CVAs and TIAs and can be classified as early and late depending on time of occurrence (less than 7 days or more than 7 days, respectively) following stroke (or TIA). Two or more recurrent late post-stroke seizures (PSS) are referred to as post-stroke epilepsy (PSE) (Slapo et al., 2006), although some have defined PSE also as first unprovoked seizure caused by a previous stroke (Jungehulsing et al., 2013). PSS can be focal or generalized, and status epilepticus can occur. PSS have a negative effect on outcome in patients with CVAs. PSS may exacerbate secondary cerebral injury by inducing glutamate excitotoxicity, and enhancing the mismatch between energy supply and demand under ischaemic conditions, leading to breakdown of ion gradients, mitochondrial damage, and eventually an irreversible state of injury (Menon and Shorvon, 2009). Experimental studies suggest that repeated seizure-like activity in the context of cerebral ischaemia significantly increases stroke size and can impair functional recovery. In people, the incidence of early and late PSS ranges from 2% to 16%, depending on study population, stroke subtype, follow-up duration and how the authors have defined early and late PSS and PSE (Arntz et al., 2013; Conrad et al., 2013; Jungehulsing et al., 2013). Patients with intracerebral haemorrhage have the highest incidence of PSS, followed by patients with ischaemic stroke and patients with a transient ischaemic attack (Arntz et al., 2013). The overall incidence of PSE is 2–6.4% in people with CVA (Arntz et al., 2013; Graham et al., 2013). In one study, the incidence of PSE was estimated as 1.2%, 3.5%, 9.0% and 12.4% at 3 months, 1, 5 and 10 years post-CVA, respectively (Graham et al., 2013). Stroke severity is a major risk factor for the developement of PSS and PSE (Arntz et al., 2013; Conrad et al., 2013; Graham et al., 2013; Jungehulsing et al., 2013).
Data on incidence of PSS and PSE in veterinary medicine are limited. In one study including 33 dogs with brain infarction, two (6%) dogs with forebrain ischaemic infarcts developed recurrent generalized seizures at 10 and 31 weeks after the diagnosis of brain infarction (Garosi et al., 2005a). In another study including 27 dogs with a clinical diagnosis of cerebral ischaemic stoke (Gredal et al., 2013), seizures were reported as part of the acute symptomatology in 15 dogs (56%). Seven of these 15 dogs developed PSE. PSE has also been reported in four of five dogs (Paul et al., 2010), 1 of 16 dogs (Gonçalves et al., 2011) with ischaemic or haemorrhagic strokes affecting the prosencephalon. In addition, recurrent seizures have been reported in 20% (15/75) of dogs with an MRI diagnosis of intracranial haemorrhage (Lowrie et al., 2012).
PSE develops more often in people with initial late PSS than in those with initial early PSS. This may be due to different pathophysiology of early and late PSS. Early PSS may be due to acute cellular biochemical disturbances either in the brain or systemically, such as altered electrolyte and acid–base balance, brain oedema, and release of excitatory neurotransmitters secondary to cerebral hypoxia or metabolic changes, whereas late PSS and PSE may result from gliotic scarring causing persistent changes in the cell networks (Slapo et al., 2006; Menon and Shorvon, 2009).
Acute ischaemia has been shown to lead to increased extracellular concentrations of glutamate and reduced GABA-ergic function, and also to functional or structural impairment of GABA-ergic interneurons. The ischaemic penumbra of a stroke (Fig. 5.5) can contain electrically irritable tissue that provides a focus for seizure activity. The area has been shown to exhibit enhanced release of excitotoxic glutamate, ionic imbalances, breakdown of membrane phospholipids and release of free fatty acids. Epileptogenesis may result from selective neuronal cell death and apoptosis, changes in cellular membrane properties, mitochondrial changes, receptor changes
(e.g. loss of GABA-ergic receptors), deafferentation and collateral sprouting (both at the site of ischaemia as well as in remote areas) and inflammatory changes. Experimental data also suggest that epileptogenesis is enhanced by hyperglycaemia at the time of ischaemia (Menon and Shorvon, 2009). Further studies are needed to clarify the pathophysiology

Fig. 5.5. Illustration of the core and penumbra of an ischaemic infarct in the brain. In the core of the ischaemic infarct, hypoperfusion is severe and results in necrosis rapidly. The degree of ischaemia and subsequent cellular damage is less severe and potentially reversible in the penumbra (which is the area surrounding the core). The brain tissue within the penumbra may recover normal cellular function if perfusion is restored promptly or may become permanently damaged if ischaemia persists.
of PSS and PSE. PSS may also occur due to recurrent strokes.
There are no evidence-based guidelines for the treatment of PSS and PSE in people and animals. In general, early PSS and particularly status epilepticus are treated aggressively (see Chapter 24). In people, recurrent early seizures are commonly treated with AEM for 3–6 months only, whereas PSE treatment is prolonged similarly to other causes of structural epilepsy (Menon and Shorvon, 2009). The choice of the AEM is influenced by the presence of concurrent disorders (e.g. renal or hepatic dysfunction), pharmacokinetic interactions with other treatments, tolerability and potential adverse effects. To date, no AEM has been identified to be clearly superior in the treatment of PSS and PSE. In people, levetiracetam is considered both safe and effective against post-stroke seizures, and may have neuroprotective effect in brain ischaemia (Belcastro et al., 2011). The benefits of neuroprotective and prophylactic antiepileptic treatment for PSE require further investigations.
Diagnostic investigations
Imaging studies of the brain such as computed tomography (CT) and magnetic resonance imaging (MRI) are necessary to support the diagnosis of CVA, to differentiate between ischaemic and haemorrhagic CVA, and to determine the location and extent of the lesion. CT is very sensitive at detecting acute haemorrhage which appears hyperdense, but it may not detect acute ischaemia in the brain (Garosi, 2010). Conventional MRI can help detecting both ischaemic and haemorrhagic CVA, however differentiation between CVA and other intracranial diseases may be challenging in some cases (Cervera et al., 2011; Wolff et al., 2012). Sensitivity and specificity of routine (not including T2* gradient echo sequences and diffusion weighted images) high-field MRI (with or without provision of clinical data) in overall lesion detection and differentiation of CVAs from neoplastic and inflammatory brain disorders in dogs are 39% and 98%, respectively. Sensitivity and specificity of routine high-field MRI (with knowledge of clinical data) are 33% and 89%, respectively, in the diagnosis of haemorragic CVAs, and 67% and 100%, respectively, in the diagnosis of ischaemic CVAs (Wolff et al., 2012). MRI pulse sequences such as T2* gradient echo (for haemorrhagic CVAs), diffusion and perfusion weighted images (for ischaemic CVAs) and magnetic resonance angiography improve the sensitivity and specificity of the diagnosis of peracute and acute CVA (Garosi, 2010; Cervera et al., 2011).
The MRI features of ischaemic CVA include an intraparenchymal lesion within a vascular territory which:
Structural Epilepsy
• shows variable contrast-enhancement (usually minimal, peripheral or heterogeneous) 7 to 10 days after onset of neurological signs.
The MRI features of haemorrhagic CVA vary depending on several intrinsic (time from ictus, oxygenation state of haemoglobin, source, size and location of haemorrhage) and extrinsic (pulse sequence and field strength) factors (Table 5.1) (Bradley, 1993; Thomas et al., 1997). Haemorrhage in areas with high ambient oxygen (ventricles; epidural, subdural and subarachnoid space) ‘ages’ more slowly than parenchymal haemorrhage, with a resultant change in time course of haemoglobin degradation. Contrast enhancement due to neovascularization in the surrounding brain tissue can occur 7–14 days after intraparenchymal haemorrhagic CVA and may be minimal, heterogenous or peripheral to ring-like.
Cerebrospinal fluid (CSF) analysis in animals with CVA is either normal, or shows aspecific changes such as mild mononuclear or neutrophilic pleocytosis, elevated protein concentration and xanthocromia. CSF should not be collected in animals with coagulopathy or increased ICP.
Once a probable diagnosis of CVA has been achieved (based on clinical presentation, MRI of the brain, and, if possible, CSF analysis), further diagnostic investigations should be performed in attempt to identify an underlying cause of ischaemic or haemorrhagic CVA (Box 5.2).
A definitive diagnosis of CVA can be reached histopathologically (Plate 3).
Treatment
Treatment of CVA focuses on prevention of secondary brain injury or complications, such as increased ICP (see management of traumatic brain injury) or seizures, and on the underlying disease. For further details on all the aspects of CVA treatment the reader is directed to more comprehensive descriptions (Garosi, 2010). Anti-epileptic treatment is performed as for other types of structural brain disorders (see section on post-stroke seizures and epilepsy, introduction to this chapter and Chapters 12–24).
Prognosis
Most dogs recover within weeks after the onset of ischaemic CVA with only supportive care (Garosi et al., 2005a). Prognosis depends on the severity of the neurological dysfunction, occurrence of complications and the underlying cause of CVA, if identified. In a retrospective study on 33 dogs with MRI or histologic diagnosis of ischaemic CVA, no association was identified between type (lacunar or territorial) or location (telencephalic, thalamic/midbrain, cerebellar) of infarct and patient outcome. Dogs with concurrent medical conditions had significantly shorter survival times than those with no identifiable medical condition and were significantly more likely to suffer from
Table 5.1. Change of appearance of intracranial haemorrhage over time (Bradley, 1993; Thomas et al., 1997).
Stage Haemoglobin state Intensity on T1WIa Intensity on T2WIa Intensity on T2*GEa
Hyperacute (<1 day) Acute
(1–3 days) Early subacute (4–7 days)
Late subacute (8–14 days)
Chronic (>14 days)
Intracellular oxyhaemoglobin
Intracellular deoxyhaemoglobin
Intracellular methaemoglobin with intact erythrocytes
Extracellular methaemoglobin with erythrocyte lysis
Ferritin and haemosiderin
| Isointense | Slightly hyperintense | Hypointense |
| Iso- to hypointense | Hypointense | Hypointense |
| Hyperintense | Hypointense | Hypointense |
Hyperintense Hyperintense Hypointense
Hypointense Hypointense Hypointense
WI: Weighted image, GE: gradient echo aIntensity is compared with normal cerebral grey matter
Box 5.2. Diagnostic investigations to identify the underlying aetiology of ischaemic or haemorrhagic CVA.
Diagnostic investigations to identify the underlying aetiology of ischaemic CVA:
recurrent neurologic signs because of subsequent infarcts. In a retrospective study on 75 dogs with an MRI diagnosis of nontraumatic intracranial haemorrhage, outcome was poor in the majority of dogs with hypertension (Lowrie et al., 2012).
Post-stroke epilepsy has a negative effect on stroke recovery and quality of life in people (Arntz, 2013).
Inflammatory/infectious
Inflammatory disease of the CNS can be classified as infectious (when caused by a known or suspected infectious agent) or immune-mediated (when the underlying aetiology is unknown and an immune-mediated process is suspected). A degree of CNS inflammation may occur also with brain neoplasia, infarction or trauma. This section will focus on infectious and presumed immune-mediated disorders. Inflammatory disease of the CNS may affect the brain parenchyma (encephalitis), the meninges (meningitis) and the spinal cord (myelitis). Inflammatory CNS disorders commonly affect young to middle-aged animals, although animals of any age and gender can be affected. Certain infectious disorders occur exclusively or predominately in certain geographic areas (Nghiem and Schatzberg, 2010). The majority of reported inflammatory CNS diseases in dogs and cats are outlined in Tables 5.3, 5.4, 5.6, 5.7, 5.8, 5.10 and 5.11. Additional information is provided in the text for selected disorders. The suggested references should be consulted for more detailed information on each disorder.
Clinical signs
Signs of systemic involvement may (e.g. certain viral diseases or mycotic disorders) or may not (e.g. cerebral abscess, neurotropic infections, immune-mediated encephalitis) be present. Ophthalmologic examination may reveal fundic changes or uveitis.
Neurological signs in animals with inflammatory CNS disease often have an acute to subacute onset, are progressive and reflect multifocal or diffuse involvement of the CNS. However, focal neurological deficits can also occur (e.g. cerebral abscess, fungal granuloma). Seizures commonly occur in animals with forebrain involvement.
Encephalitis-related seizures and post-encephalitic epilepsy
Seizures can occur during the active stage of cerebral inflammation, disappear after the inflammation/infection has resolved, they may persist, or they may first manifest in the post-encephalitic period after the inflammation/infection has resolved (see Schwartz-Porsche and Kaiser, 1989; Michael and Solomon, 2012; Chapter 3). The exact risks of developing encephalitis-associated seizures are poorly understood, but appear to relate to
Structural Epilepsy
the pathogen, the degree of cortical involvement and the cytokine-mediated inflammatory response (Michael, 2012).
Acute post-encephalitic seizures (PES), defined by the International League Against Epilepsy (ILAE) as seizures occurring within 7 days of an acute central nervous system (CNS) infection, have been reported in 2–67% of patients with encephalitis. Late (>7 days) PES usually develop within the first 5 years following encephalitis, but can occur up to 20 years later. The pathogen causing the encephalitis appears important in predicting the likelihood of later developing post-encephalitic epilepsy (PEE). Studies from Western industrialized countries show that patients with encephalitis are overall about 16 times more likely than the general population to develop late PES (Michael and Solomon, 2012). Data on incidence and risk factors for encephalitis-associated seizures are unavailable in veterinary medicine.
Several potential pathophysiologic mechanisms can explain the development of seizures in patients with encephalitis. Experimental evidence indicates a significant role for inflammatory and immune mediators in initiation of seizures and epileptogenesis (Friedman and Dingledine, 2011; Kramer et al., 2012). The inflammatory response and in particular inflammatory mediators (including cytokines such as interleukins (IL), chemokines, prostaglandins and complement factors) produced by astrocytes and microglia are increasingly recognized to promote excitatory neurotransmitter release and consequent depolarization. These inflammatory mediators can have both acute and long-term effects on seizure threshold (Vezzani et al., 2011; Michael and Solomon, 2012). Glial cells, as well as neurons, can over-express receptors for inflammatory molecules, including receptors for proinflammatory cytokines
(e.g. IL-1β, IL-6 and tumour necrosis factor-α), as well as toll-like receptors. Cytokines or prostaglandins can induce post-translational changes in receptor-coupled or voltage-dependent ion channels leading to increased glutamatergic neurotransmission or reduced GABA-mediated effects. Proinflammatory cytokines also can decrease glutamate reuptake by astrocytes and can increase the release of excitatory gliotransmitters by activated glial cells, possibly also contributing to neuronal network hyperexcitability. Long-term effects of inflammatory mediators involve gene transcription of proinflammatory genes, which may perpetuate inflammation in brain tissue, and play a role in alterations in blood–brain barrier (BBB) permeability properties. A compromised BBB in turn contributes to decrease seizure threshold by inducing ionic imbalance in the extracellular milieu, as well as astrocytes and microglia dysfunctions. In addition, recent data suggest that cytotoxic T-cells and antibody-mediated complement activation may have a role in neural tissue degeneration and subsequently epileptogenesis (Bauer et al., 2012).
In people, there is currently no evidence to support prophylactic anti-epileptic treatment in all patients with encephalitis. Patients with encephalitis should be closely monitored and administed AEMs promptly if seizures occur. PEE should be treated similarly to other types of structural epilepsy. People with PEE are frequently refractory to AEMs and may require combination therapy or neurosurgery to attempt to control seizures (Michael and Solomon, 2012).
Diagnostic investigations
Haematology may sometimes provide evidence of systemic infection (e.g. alterations in white blood-cell count), and serum biochemistry may reveal changes consistent with involvement of other organs. Cerebrospinal fluid analysis often reveals increased white blood-cell (WBC) count (pleocytosis) and increased protein concentration. CSF pleocytosis has been classified as mild (6–50 WBC/µl), moderate (51–200 WBC/µl), or marked (>200 WBC/µl), and as mononuclear, neutrophilic, eosinophilic, or mixed based on the predominant cell type on cytological examination (Tipold, 2003).
The type of CSF pleocytosis may be suggestive of a particular aetiology or disease group (Table 5.2).
CSF may be normal when the CNS inflammation does not involve the leptomeninges or the ependymal lining of the ventricular system or if the animal has been treated with anti-inflammatory medications (particularly corticosteroids) prior to CSF
Table 5.2. Cerebrospinal fluid characteristics of canine and feline inflammatory CNS disease.
Total protein Disease Type of pleocytosis concentration
| Viral meningoencephalitis | None or mild to moderate mononuclear | Normal to |
| (CDV or other; FIP | markedly | |
| excluded) | elevated | |
| FIP meningoencephalitis | Moderate to marked neutrophilic or mixed, | Markedly elevated |
| occasionally eosinophilic | ||
| Rickettsial | Mild to moderate mononuclear or mixed. Can be | Mildly to markedly |
| meningoencephalitis | neutrophilic with granulocytic ehrlichiosis or | elevated |
| anaplasmosis | ||
| Bacterial meningoencephalitis | Moderate to marked neutrophilic (toxic changes in | Mildly to markedly |
| cell morphology) in acute and subacute | elevated | |
| infections; mixed in chronic infections; sometimes | ||
| mononuclear following antibiotic treatment | ||
| SRMA | Moderate to marked neutrophilic in acute SRMA, | Mildly to markedly |
| mononuclear or mixed in chronic SRMA | elevated | |
| Protozoal meningoencephalitis | Moderate mixed, occasionally eosinophilic, | Mildly to markedly |
| rarely mononuclear | elevated | |
| Fungal meningoencephalitis | Moderate to marked mixed, occasionally | Markedly elevated |
| eosinophilic | ||
| Algal (Prototheca) | Moderate to marked mixed or eosinophilic | Markedly elevated |
| Parasitic | Mild to moderate mixed, often eosinophilic | Mildly to markedly |
| meningoencephalitis | elevated | |
| GME | None or moderate to marked mononuclear | Mildly to markedly |
| or mixed | elevated | |
| Necrotizing | Mild to marked mononuclear | Mildly elevated |
| meningoencepahlitis/ | ||
| leukoencephalitis | ||
| Eosinophilic | Mild to marked eosinophilic | Mildly to markedly |
| meningoencephalitis | elevated |
collection. Additional tests on CSF (such as polymerase chain reaction (PCR), antibody or antigen titres, immunofluorescence and culture) can help to reach an aetiologic diagnosis. Occasionaly, certain microorganisms (e.g. bacteria, Ehrlichia morulae, fungi, protozoa or parasites) can be visualized on CSF cytology.
Inflammatory CNS disease can cause increased intracranial pressure (ICP) and CSF collection may be contraindicated due to the risk of cerebral herniation and death. Progression from obtundation to stupor, a diminished or absent vestibulo-ocular reflex, the development of unilateral or bilateral midriasis and loss of the pupillary light reflexes are suggestive of increased ICP and transtentorial brain herniation. Any time increased ICP is suspected, MRI of the brain should be performed before considering CSF collection.
MRI of the brain can reveal changes suggestive of inflammatory CNS disease such as multifocal, diffuse or sometimes focal lesions within the brain parenchyma that typically appear hyperintense on T2-weighted and FLAIR images, iso- to hypointense on T1weighted images and show variable contrast enhancement sometimes with meningeal involvement following administration of contrast medium. The MRI findings sometimes may support the ante-mortem diagnosis of a particular aetiology (e.g. FIP). The MRI features of various inflammatory CNS diseases have been reviewed (Hecht and Adams, 2010b). Sensitivity and specificity of high-field MRI for classifying brain diseases as inflammatory are 86.0 and 93.1% without provision of clinical data and 80.7 and 95.4% with provision of clinical data, respectively (Wolff et al., 2012). Sensitivity and specificity of high-field MRI
Structural Epilepsy
are lower for detection of specific inflammatory CNS aetiologies (Wolff et al., 2012).
Treatment
Treatment of CNS infections depends on the identification of the aetiologic agent and selection of the appropriate antimicrobial or anti-fungal agent (see Tables 5.5–5.9). Treatment of viral CNS infections is mostly supportive and symptomatic. Treatment of non-infectious inflammatory CNS disorders involves immunosuppressive medications. Anti-epileptic treatment of both acute and late PES is performed as for other types of structural brain disorders (see section on encephalitic-related seizures, introduction to this chapter and Chapters 12–24). Interactions between AEMs and other medications to treat the CNS infection/ inflammation should be carefully considered. In animals with increased intracranial pressure secondary to CNS inflammation, treatment and monitoring should be performed similarly to what has been described for animals with traumatic brain injury (see management of traumatic brain injury).
Prognosis
Prognosis is variable and dependent on the underlying aetiology, extent and severity of the CNS inflammation and associated neurological deficits, and promptness of diagnosis and treatment (when available).
Canine distemper
Canine distemper (CD) is a common polysystemic disease of dogs that may infect the CNS. CD is caused by canine distemper virus (CDV), an RNA virus that belongs to the genus Morbillivirus, family Paramyxoviridae (Martella et al., 2008; Greene and Vandevelde, 2012). Although the incidence of CD has decreased since the introduction of the modified-live CDV vaccines in the 1950s, it is still a common CNS disorder worldwide, primarily in unvaccinated dogs but also occasionally in dogs with a vaccinal history as immunity to virulent CDV is not absolute after vaccination. Dogs that are not immunized regularly may lose their protection and become infected following stress, immunosuppression or contact with diseased animals. CDV is quickly inactivated in the environment and transmission mainly occurs by direct animal-to-animal contact or by exposure to infectious aerosol. The virus enters the new host by the nasal or oral route and promptly starts replication in the lymphoid tissues (Martella et al., 2008). Eight to 9 days after infection, CDV spreads by cell-associated viraemia to the epithelial cells in most organs and CNS tissue. Transient fever, loss of appetite, lethargy, ocular and nasal discharge and tonsillitis may be observed. At this stage, the outcome of the infection and the severity of the clinical signs vary markedly depending on strain virulence, environmental conditions and the dog’s age and immune status. If the dog develops a strong immune response, the virus is cleared from most tissues and the animal shows no clinical signs of infection. If the immune response is intermediate by day 9 to 14 post-infection, the virus is able to reach the epithelial tissues (resulting mainly in respiratory and gastrointestinal signs) and the CNS. The respiratory and gastrointestinal signs may resolve as antibody titre increases. However, CDV may persist for extended periods in the uvea, neurons, urothelium and in some cutaneous areas such as the foot pads. The CNS signs are generally delayed and hyperkeratosis is observed in some dogs. Dogs unable to mount an adequate immune response by day 9 to 14 post-infection undergo viral spread to many tissues, develop severe and rapidly progressive clinical signs and die.
Clinical signs
Several clinical syndromes associated with distemper have been recognized in dogs:
acute distemper. Acute distemper occurs in susceptible young dogs unable to mount an adequate immune response. Respiratory and gastrointestinal signs predominate and are often exacerbated by secondary bacterial infections resulting in mucopurulent conjunctivitis, chorioretinitis, rhinitis, coughing, dyspnoea, pneumonia, diarrhoea and vomiting. Neurologic signs (mainly seizures) may occur later in the clinical course but dogs may die before these
| Affected | Source of | Treatment and | ||||
| species | Disease | Virus | infection | Clinical signs | Diagnosis | prevention |
| Dog and cat Dog and cat | Rabies Pseudorabies | Rhabdovirus, genus Lyssavirus, family Rhabdoviridae Pseudorabies virus (PRV) or porcine herpesvirus-1 | Most commonly bite wounds from infected animals that are secreting virus in their saliva Rarely, wound contamination from infected saliva. Ingestion of infected raw pork meat, or parenteral inoculation (e.g. scratches, penetrating wounds) from virus-contaminated | Initially behavioural changes for 1–3 days, then progression to excitatory (‘furious’) form (restlessness, wandering, excessive salivation, pica, viciousness and seizures) and/ or to paralytic (‘dumb’) form (severe obtundation, progressive LMN paralysis from the site of viral entry to the brainstem, facial and lower jaw paralysis, pharyngeal and hypoglossal paralysis resulting in difficulty in eating and drinking, and drooling of saliva). The furious form is more common in cats and the paralytic form is more common in dogs, however the two forms often overlap in dogs Respiratory failure and death occur 3 to 10 days after onset of clinical signs. Behavioural changes, restlessness, intense pruritus and self-mutilation at site of viral entry (Fig. 5.6), excessive salivation, multiple cranial nerve deficits, ataxia, paresis, aggression, seizures; rapid progression to coma and death within 24 to 48 h after onset of clinical signs | Post-mortem by direct fluorescent antibody assay (dFA) for viral antigen or direct rapid immunohistochemistry test on brain tissue. No pre-mortem diagnostic test has adequate sensitivity to be consistently reliable for diagnosis of rabies in animals. Mononuclear pleocytosis and increased protein concentration can occur in CSF of dogs and cats with post-vaccinal rabies Post-mortem by dFA for viral antigen or PCR on brain and tonsil tissue. Mononuclear pleocytosis and increased protein concentration can occur in CSF | There is no treatment. Prevention by vaccination. Post-vaccinal rabies has rarely been reported in dogs and cats There is no treatment. Prevention: avoid contact with infected pigs or raw pork meat in endemic areas |
| objects |
116 L. De Risio
| Dogs | Central European | Arbovirus, genus |
| tick-borne | Flavivirus, family | |
| encephalitis | Flaviviridae |
| Dog and | West Nile virus | Arbovirus, |
| cat | genus Flavivirus, | |
| family | ||
| Flaviviridae |
| Dog | Canine distemper | Canine distemper |
| (see text for | virus (CDV), genus | |
| more | Morbillivirus, | |
| information) | family Paramyx | |
| oviridae |
Bites from ticks (Ixodes ricinus and Ixodes persulcatus)
Bites from ornithophilic mosquitoes, primarily of the genus Culex
Dogs and cats and other mammals are incidental or dead-end hosts.
By inhalation or ingestion
Fever and multifocal neurological signs including myoclonus, seizures, multiple cranial nerve deficits, UMN or LMN paresis, stupor. Death can occur 4–7 days after onset of neurologic signs
Fever, anorexia, lethargy, obtundation, ataxia, weakness and rarely seizures
Vary markedly depending on strain virulence, and the dog’s age and immune status. Neurological signs including seizures, myoclonus, obtundation, head tilt, nystagmus, ataxia and paresis may occur alone, in association or following respiratory, gastrointestinal and ocular signs. Enamel hypoplasia and foot pad and nose hyperkeratosis may be observed Raising antibody titres.
Post-mortem, histopathology, immunohistochemistry (IHC).
Mononuclear pleocytosis and increased protein concentration can occur in CSF
Virus-specific IgM and IgG.
Post-mortem, IHC, reverse transcriptase polymerase chain reaction (RT-PCR) on tissues, viral isolation
Reverse transcriptase polymerase chain reaction (RT-PCR) and real-time RT-PCR on blood, CSF, urine or conjunctival scraping; viral antigen detection by dFA on neural tissue, cerebrospinal fluid cells (infected lymphocytes), footpad biopsy, or other tissues; IHC of skin biopsy or brain tissue.
CDV-specific antibody ratio in CSF and serum. Intracytoplasmic inclusions in CSF mononuclear cells
Supportive treatment, glucocorticoids may worsen disease progession.
Prevention of tick bites in endemic areas
Supportive treatment.
Prevention: minimizing exposure to infected mosquitoes (insect repellants); vaccination
Supportive and symptomatic treatment. Prevention by vaccination
Continued
Structural Epilepsy
| Affected | Source of | Treatment and | ||||
| species | Disease | Virus | infection | Clinical signs | Diagnosis | prevention |
| Dog | Canine herpes virus | Canine herpes virus | In utero, by direct contact with diseased | Typically affects young puppies. Clinical signs reflect systemic involvement (diarrhoea, dyspnoea, | Viral isolation, detection of viral nucleic acid on tissues by PCR or in situ | Supportive treatment |
| litter-mates, or by inhalation or ingestion of | obtundation to coma, opisthotonus and seizures). Often fatal | hybridization | ||||
| infected material | ||||||
| Dog | Infectious canine hepatitis | Canine adenovirus | By inhalation or ingestion of | Most frequently affects young dogs. Clinical signs reflect systemic | Evidence of hepatic dysfunction on clinical | Supportive treatment. |
| type I | infected material | involvement (hyperthermia, vomiting, diarrhoea, lympho-adenomegaly, abdominal pain, hepatomegaly). | pathology profile, serology, viral isolation, immunofluorescence | Prevention by maternal immunity and | ||
| Neurological signs (obtundation to coma, seizures) may develop | vaccination | |||||
| Cat | Feline infectious peritonitis (FIP) (see text for more information) | Feline coronavirus (FCoV), genus Coronavirus (Geselavirus), | By ingestion (and rarely inhalation) of material contaminated | Neurological signs are most common with the non-effusive form of FIP and include abnormal mentation and behaviour, head tilt, seizures, ataxia (generally vestibular), cranial nerve | Detection of FCoV antigen within macrophages in body fluids, cytology or biopsy specimens by dFA or IHC | Glucocorticoids; feline interferon-ω; polyprenyl immuno |
| family Coronaviridae | by infected faeces | dysfunction, and varying degrees of proprioceptive and motor deficits. Neurological signs can occur either | stimulant (non-effusive FIP). Prevent infection | |||
| alone or in conjunction with systemic signs such as fever, vomiting, diarrhoea, anorexia, weight loss and | with FCoV (whenever possible) | |||||
| lethargy. Clinical signs are slowly progressive and often fatal in cats with CNS FIP | ||||||
| Cat | Feline leukaemia virus (FeLV) | Retrovirus, subfamily Oncornavirus | By inhalation or ingestion of infected | Systemic involvement and immunosuppression, FeLV-induced lymphoma, possible brain | ELISA, dFA, PCR | Supportive and symptomatic treatment. |
| material (mainly contaminated | involvement | Prevention by vaccination | ||||
| saliva) |
118 L. De Risio
Cat Feline immunodeficiency virus (FIV)
Cat Feline paramyxovirus
Cat Feline panleukopaenia
Cat and Borna disease dog Lentivirus
Family Paramyxoviridae, genus Paramyxovirus, Morbillivirus, Hendravirus
Feline panleukopaenia virus, Parvovirus
Borna disease virus (BDV), family Bornaviridae
By parenteral inoculation of virus present in saliva or blood by bite and fight wounds
Probably by inhalation or ingestion of infected material
In utero or early neonatal infection by exposure to contaminated material. The virus has long environmental survival
Probably by inhalation or ingestion of infected material. Viral entry into nerve endings in the olphactory, oropharyngeal or gastrointestinal mucosae
Fever, dermatitis, otitis, stomatitis, lymphoadenomegaly, enteritis, respiratory disease, ocular disease, neurological signs (behavioural changes, and rarely seizures and paresis). Clinical signs may result also from opportunistic viral, bacterial, protozoal or fungal infections
Signs of CNS infection, including seizures
Cerebellar signs due to cerebellar hypoplasia. Rarely signs of forebrain involvement including behavioural changes and seizures. Clinical signs are present at birth and are non-progressive. Presence and degree of neurological dysfunction can vary in kittens of the same litter
Cats: behavioural changes, obtundation, ataxia, pelvic limb paresis or paralysis, tremors, seizures, visual impairment, inability to retract the claws, fever and constipation.
Dogs: acute onset and rapidly progressive forebrain signs
ELISA
Post-mortem detection of viral antigen in the CNS
Clinical presentation, leukopaenia, cerebellar hypoplasia on MRI or CT, ELISA for viral antigen in faeces or intestinal content, viral isolation
Post-mortem detection in the CNS of BDV antigen by IHC and/or BVD RNA by in situ hybridization combined with histological and clinical findings of nonsuppurative meningoencephalitis
Supportive and symptomatic treatment.
Prevention by vaccination
Supportive and symptomatic treatment
Prevention by vaccination
Supportive treatment, glucocorticoids may be beneficial.
Prevention of predation on mice in endemic rural woodland
Structural Epilepsy
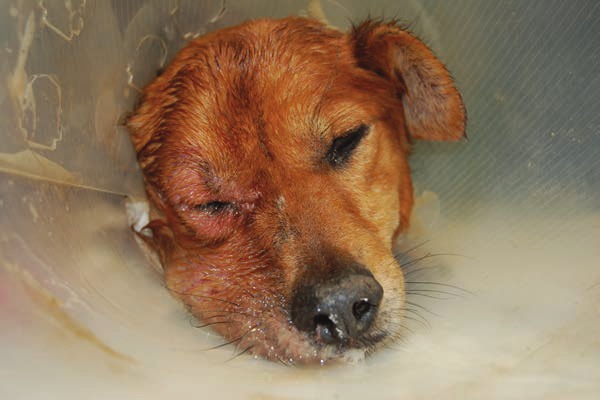
signs develop. Histological findings are consistent with a polioencephalomyelitis.
chronic distemper encephalomyelitis. Chronic distemper encephalomyelitis occurs in young dogs that survive the acute stages of the disease and in mature dogs without signs of systemic disease. Animals vaccinated against CDV may be affected. Enamel hypoplasia and hyperkeratosis of the foot pads and nose may be observed in dogs that survive subclinical or subacute infections. Neurological signs are commonly progressive and include obtundation, head tilt, nystagmus, ataxia (usually vestibular and/or cerebellar), paresis, constant repetitive myoclonus (constant repetitive sudden involuntary contractions, followed immediately by relaxation of a specific muscle group) involving head or appendicular muscles, and generalized or focal seizures. Constant repetitive myoclonus may persist while the animal is asleep. The so-called ‘chewing gum fits’ described in dogs with CDV are characterized by rhythmic biting movements of the mandible and may represent a form of focal seizures associated with temporal lobe polioencephalomalacia. Visual deficits can occur due to chorioretinitis and optic neuritis. Histological findings are characterized by severe demyelinating meningoencephalomyelitis.
Lesions are most common in the cerebellum, cerebellar peduncles, cervical spinal cord, optic tracts and periventricular white matter.
old dog encephalitis. Old dog encephalitis is a rare form of CD that appears to be a manifestation of chronic viral infection after years of latent brain infection. There are no related systemic signs. The most common initial neurological sign is visual impairment. Other neurologic signs are progressive and include personality changes, obtundation, compulsive circling and head-pressing against objects. Histological findings are characterized by perivascular mononuclear cuffs, microglial proliferation, astrogliosis, neuronal degeneration and neuronophagia mostly within the cerebral cortex. Necrosis of cerebral grey matter may be observed.
Clinical signs may also be associated with combined infections (e.g. Toxoplasma gondii or Neospora caninum).
Diagnostic investigations
Haematological and biochemical findings are nonspecific and include absolute lymphopaenia, hypoalbuminaemia and hyperglobulinaemia during the acute phase of illness. CSF analysis may reveal mononuclear pleocytosis
Structural Epilepsy
(>5 WBC/µl) and elevated protein concentration (>25 mg/dl). Eosinophilic intracytoplasmic inclusions may be found in CSF or peripheral blood cells although their detection is rare. CSF can be normal in dogs with acute noninflammatory demyelinating encephalomyelitis. A CDV-specific antibody ratio (CDVspecific IgG in CSF/CDV specific IgG in serum) higher than canine adeno- or parvovirusspecific antibody ratio (IgG in CSF/IgG in serum) can help to identify intrathecal production of CDV-specific IgG (see antigen-specific antibody index, Chapter 10). MRI of the brain may be normal or may reveal lesions that are hyperintense on T2WI, isointense or hypointense on T1WI with inconsistent contrast enhancement, and loss of cortical grey/white matter demarcation. The lesion distribution varies depending on the stage of CDV encephalitis (Bathen-Noethen et al., 2008; Griffin et al., 2009).
The diagnosis of CD can be achieved by molecular assays, such as reverse transcriptase polymerase chain reaction (RT-PCR) and real-time RT-PCR on blood, CSF, urine, conjunctival swabs or tissue specimens (Elia et al., 2006; Saito et al., 2006), as well as detection of viral antigen by direct fluorescent antibody assay (dFA) (on neural tissue, cerebrospinal fluid cells (infected lymphocytes), footpad biopsy, or other tissues) or by immunohistochemistry (IHC) on biopsy specimens
(e.g. nasal mucosa, foot pad, haired skin of the dorsal neck) or post-mortem.
Treatment
Treatment consists of supportive care and antibiotics and is aimed at preventing the secondary bacterial infections that are frequent in immunosuppressed animals. Anti-epileptic treatment is performed as for other types of structural brain disorders (see introduction to this chapter and Chapters 12–24). Seizures secondary to CDV encephalitis have been reported to be difficult to control with phenobarbital (Tipold et al., 1992).
Modified-live vaccines are recommended for immunization of dogs to prevent CDV.
Although vaccine-induced disease is always suspected in dogs that develop distemper shortly after immunization, in most cases, the disease is induced by wild-type CDV infecting pups before active immunization is elicited (Martella et al., 2008).
Prognosis
Prognosis is variable depending on the clinical syndrome associated with CDV infection and generally varies from persistent neurological deficits (particularly myoclonus) to death.
Feline infectious peritonitis
Feline infectious peritonitis (FIP) and viral non-FIP encephalitides (i.e. non-suppurative encephalitides of unknown, although probable viral, aetiology based on histological findings) are the two most commonly recognized infectious CNS disorders of cats (Gunn-Moore and Reed, 2011).
FIP is a highly fatal, progressive and immune-augmented disease of cats caused by infection with feline coronavirus (FCoV). Although the terms feline infectious peritonitis virus (FIPV) and feline enteric corona-virus (FECV) have been used to refer to the virus causing FIP and the ubiquitous benign enteric virus, respectively, the term FCoV should be used to describe all coronaviruses in cats. FCoV is an RNA virus and belongs to the genus Coronavirus of the family Coronaviridae. It has been proposed that FCoV along with swine and canine coronaviruses becomes part of a new species called Geselavirus (Addie, 2012). FCoV causes a ubiquitous enteric infection in cats, which leads to FIP in approximately 1–3% of cats. The etiopathogenesis of FIP is complex and still unclear. A widely cited pathogenetic hypothesis is the ‘in vivo mutation transition hypothesis’ also called the ‘internal mutation hypothesis’. This postulates that viral mutations occur in healthy FCoV-infected cats giving rise to virulent virions that are able to replicate within macrophages and disseminate systemically leading to FIP (Pedersen, 2009). The precise nature of the mutation responsible for this pathogenetic hypothesis has not been identified yet. An alternative ‘circulating avirulent and virulent FCoV hypothesis’ suggests that distinctive benign and pathogenic strains of FCoV circulate in a population, and susceptible individuals exposed to the virulent strains develop FIP (O’Brien et al., 2012). It is likely that FIP etiopathogenesis is more complex than either hypothesis alone would suggest.
FCoV has a worldwide distribution and therefore cats worldwide are susceptible to developing FIP (Kent, 2009). FCoV is endemic especially in environments in which many cats are kept together in a small space (e.g. catteries, shelters, pet stores). Although cats of any age can develop FIP, the risk is higher in kittens and cats up to 2 years of age or older than 10 years of age. The risk to develop FIP appears greater in young and immune-compromised cats as well as in cats with a high viral load (Hartmann, 2005). In addition, pure-breed cats (such as the Abyssinian, Bengal, Birman, Himalayan, ragdoll, rex) have a greater risk of developing FIP than non-pure-breed cats (Pesteanu-Somogyi et al., 2006). Infection usually takes place by ingestion (or, rarely, by inhalation) of material contaminated by infected faeces shed by a cat with FCoV infection or by a cat with FIP (Hartmann, 2005). Many healthy cats shed FCoV intermittently or continuously for up to 10 months post-infection or longer, serving as chronic carriers and thereby perpetuating reinfection of other cats (Hartmann, 2005).
Clinical signs
Clinical signs of FIP can be variable because many organs can be involved. Three different forms of FIP have been identified: (i) an effusive (also called exudative or wet) form characterized by abdominal, thoracic and pericardial effusions; (ii) a non-effusive (also called granulomatous, non-exudative, dry or parenchymatous) form characterized by granulomatous changes in different organs, including the CNS, the eyes, kidneys, mesenteric lymph nodes, bowel wall and liver; and
(iii) a mixed form. The effusive and non-effusive forms can transform into each other and should be considered the gradations of the same process characterized by pyogranulomatous vasculitis (Hartmann, 2005; Addie, 2012). Neurological signs are most common with the non-effusive form of FIP and can occur either alone or in conjunction with systemic signs such as fever, vomiting, diarrhoea, anorexia, weight loss and lethargy. Neurological signs include abnormal mental status and behaviour, head tilt, seizures, ataxia (generally vestibular), nystagmus cranial nerve dysfunction, and varying degrees of proprioceptive and motor deficits. Seizures can be generalized tonic-clonic or focal, and status epilepticus can occur (Timmann et al., 2008). Ocular signs of FIP comprise anterior uveitis (often with keratic precipitates) (Plate 4), chorioretinitis, anisocoria and retinal haemorrhage, detachment and cuffing of the retinal vasculature. Clinical signs are slowly progressive and eventually fatal.
Diagnostic investigations
Definitive diagnosis of FIP ante-mortem is challenging due to the nonspecific clinical signs, lack of pathognomonic haematologic and biochemical abnormalities and low sensitivity and specificity of tests routinely used in practice (Hartmann, 2005). Haematology usually reveals a normocytic, normochromic, non-regenerative anaemia, neutrophilic leukocytosis and lymphopaenia. Approximately 50% of cats with the exudative form and 70% of cats with the granulomatous form of FIP have increased serum proteins, primarily due to hyperglobulinaemia. Protein electrophoresis reveals a polyclonal gammopathy, mainly involving the γ-globulins. Other serum biochemical changes may be observed depending on the severity of involvement of other organ systems including abnormal hepatic enzyme, bilirubin, urea nitrogen and creatinine levels. When present, effusions should be analysed and typically they are consistent with a modified transudate (protein content >3.5 g/dl, cellular content <5000 nucleated cells/ml) in cats with FIP. An effusion with protein content >3.5 g/dl, albumin/globulin ratio <0.45, and low cellulatiry consisting predominantly of neutrophils and macrophages is highly predictive of FIP (Addie, 2012). In cats with neurological signs, CSF analysis may reveal elevated protein (50–350 mg/dl) and pleocytosis (100–10,000 WBC/µl) containing mainly neutrophils, lymphocytes and macrophages. CSF collection may be difficult or impossible
Structural Epilepsy
due to the high viscosity of the fluid caused by the high protein and inflammatory cell content, therefore additional care should be taken when collecting CSF in cats with suspected FIP and ideally MRI should be performed before attempting CSF collection. In cats with CNS, FIP, MRI may show T2 and FLAIR hyperintensity and contrast enhancement of ventricular lining, choroid plexus and meninges compatible with ependymitis, choroiditis and meningitis (Fig. 5.7a–e). Concurrent hydrocephalus is common, and herniation of the cerebellum secondary to increased intracranial pressure is possible (Negrin et al., 2007). Detection of FCoV antigen within macro-phages in body fluids, as well as in cytologic or biopsy specimens by direct immunofluorescence or immunohistochemistry confirms the diagnosis of FIP, however false negatives can occur (Hartmann, 2005). RT-PCR identification of FCoV in blood or effusions does not provide a definitive diagnosis of FIP and RT-PCR on CSF has low sensitivity (31% in one study) (Foley et al., 1998). Quantitative RT-PCR may help to increase sensitivity and specificity in the diagnosis of CNS FIP (Nghiem and Schatzberg, 2010). Characteristic histopathologic CNS lesions of FIP are a pyogranulomatous meningoencephalitis and lymphoplasmacytic periventriculitis.
Treatment
Treatment is aimed at suppressing the inflammatory and immune-mediated response of the cat’s immune system to virulent FCoV. Treatment protocols include glucocorticoids and feline interferon-ω for effusive FIP, and polyprenyl immunostimulant (or alternatively glucocorticoids) and feline interferon-ω for non-effusive FIP (Addie, 2012).
Prognosis
Prognosis is guarded to poor in cats with effusive FIP and in cats with non-effusive FIP resulting in neurologic signs. Prevention of FIP should focus on preventing infection with FCoV (whenever possible). Safety and efficacy of vaccination requires further evaluation (Hartmann, 2005; Addie, 2012).
Bacterial diseases of the CNS in dogs and cats
Bacterial diseases of the CNS can be caused by aerobic and anaerobic organisms (Table 5.4) and can result in meningitis, encephalitis, myelitis, abscessation (focal or multifocal) or empyema.
Multiple organism infections can occur. Bacterial CNS infections are relatively uncommon in dogs and cats. Bacteria can gain access to the CNS haematogenously from a distant septic focus (e.g. endocarditis, urinary tract infections, pulmonary infections), by extension of infection from structures adjacent to the nervous system, such as the nasal passages, sinuses, internal ears, dental roots and eyes, or by direct penetration into the CNS such as occurs with bite wounds, migrating plant foreign bodies, and previous trauma or surgery (Radaelli and Platt, 2002; Dennis et al., 2005; Sturges et al., 2006; Kent, 2012). A compromised immune system can predispose to bacterial colonization of the CNS (Smith et al., 2007).
Table 5.4. Bacterial diseases of the CNS in dogs and cats.
Gram Organisms reaction Shape
Aerobic/facultative anaerobic organisms
| Staphylococcus spp. | Positive | Coccus |
| Streptococcus spp. | Positive | Coccus |
| Corynebacterium spp. | Positive | Rod |
| Pasturella spp. | Negative | Rod |
| Escherichia coli | Negative | Rod |
| Proteus spp. | Negative | Rod |
| Pseudomonas spp. | Negative | Coccus |
| Salmonella spp. | Negative | Rod |
| Klebsiella sp. | Negative | Rod |
| Bartonella spp. | Negative | Rod |
| Brucella canis | Negative | Coccus |
| Nocardia spp. | Positive | Rod |
| Other aerobic organisms | ||
| Anaerobic organisms | ||
| Bacteroides spp. | Negative | Rod |
| Fusobacterium spp. | Negative | Rod |
| Peptostreptococcus spp. | Positive | Coccus |
| Eubacterium spp. | Positive | Coccus |
| Actinomyces spp. | Positive | Rod |
| Other anaerobic | ||
| organisms |


Fig. 5.7. MRI of the brain of a 4-month-old, male, domestic short hair cat with FIP resulting in multifocal CNS signs and mild abdominal effusion. Transverse FLAIR images at the level of caudate nucleus (a), pons (b) and medulla oblongata show hyperintensity of the lining (ependyma) of both lateral ventricles (a), meninges (b, c) and choroid plexus of the fourth ventricle (c). Sagittal T1W (d) and T1WC (e) images of the caudal fossa and adjacent regions. Note the diffuse meningeal enhancement around the brain stem and of the ependymal lining of the dilated fourth ventricle.
Structural Epilepsy
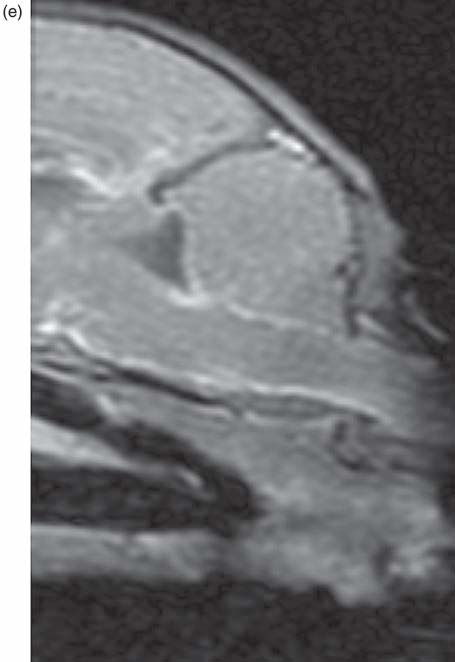
Clinical signs
General physical examination in animals with bacterial CNS infection may be normal or reveal fever and the involvement of other organs such as the lungs, gastrointestinal tract, urinary tract, prostate, skin and ears. Severely systemically affected animals may present with hypovolaemic shock and signs of disseminated intravascular coagulation. Animals with cerebral abscessation may show signs of a previous bite wound, trauma or surgery on the head.
Neurological signs vary depending on the location and extent of the infection and associated inflammation and include spinal hyperalgesia, behavioural or personality changes, altered mental status, ataxia, paresis, postural reaction deficits, cranial nerve dysfunction and seizures (Radaelli and Platt, 2002). Onset of signs is generally acute and rapidly progressive.
Fever and cervical hyperalgesia have been reported in about 40% and 30% of dogs with bacterial meningoencephalomyelitis, respectively (Radaelli and Platt, 2002). Seizures have been reported in 11 to 30% of dogs with bacterial meningoencephalomyelitis (Radaelli and Platt, 2002). Death may occur due to brain herniation and acute respiratory insufficiency in animals with severe brain oedema or mass effect.
Diagnostic investigations
Haematology may reveal a systemic inflammatory response. Abnormalities including neutrophilic leucocytosis with or without a left shift or leukopaenia, and thrombocytopaenia have been reported to occur in about 57% of dogs with bacterial meningoencephalomyelitis (Radaelli and Platt, 2002). Haematological abnormalities appear uncommon in cats with cerebral abscess (Costanzo et al., 2011).
Serum biochemistry abnormalities may be present and reflect involvement of other organs.
MRI or CT of the brain may reveal various parenchymal and meningeal signal changes in animals with meningoencephalitis, a space-occupying mass lesion in animals with cerebral abscessation as well as abnormalities of contiguous structures (e.g. otitis mediainterna, sinusitis, retrobulbar mass) representing the source of the infection. The CT and MRI characteristics of cerebral abscessation can change over time reflecting the different stages of abscess development (Costanzo et al., 2011). MRI findings broadly include a mass lesion that is hypointense on T1WI, hyperintense on T2WI and FLAIR, contrast enhances (with a peripheral ring pattern in mature abscess) and causes perilesional white matter oedema (often severe) and mass effect on adjacent brain parenchyma potentially resulting in subfalcine, caudal subtentorial and/or foramen magnum herniation (Fig. 5.8a–f). A skull defect and subcutaneous soft tissue changes may be detected at the site of the penetrating head injury (Costanzo et al., 2011). The CT appearance of well-encapsulated abscesses shows a ring-shaped contrast-enhancing lesion.
As with any disorder in which raised intracranial pressure is suspected, CSF collection should be performed only if MRI shows no significant risk of brain herniation and CNS parenchymal damage. In animals with bacterial CNS infection intracranial pressure may be raised secondary to diffuse parenchymal oedema or due to a focal lesion (such as an abscess) resulting in severe mass effect on adjacent brain parenchyma. CSF analysis in animals with acute and subacute meningoencephalitis or meningoencephalomyelitis typically reveals marked neutrophilic pleocytosis (WBC >500/µl) with toxic changes in cell morphology and severely increased protein concentrations. Mixed pleocytosis may occur with chronic infection and mononuclear pleocytosis may be observed following treatment. In a study in dogs with bacterial meningoencephalomyelitis, the mean protein concentration was 337.0 mg/dl (reference range 15.0–35.0 mg/dl) and 93% of samples exhibited pleocytosis (Radaelli and Platt, 2002). Rarely, intracellular bacteria may be seen during CSF cytological examination.
Aerobic and anaerobic cultures of CSF, blood, urine or material from a septic focus (including a cerebral abscess or middle ear infection) may allow identification of the causative bacterial organism and determination of its antibiotic susceptibility. However, CSF, blood and urine culture results have been reported to be negative in approximately 80% of dogs with confirmed bacterial meningoencephalomyelitis. PCR using broad-range primers can help to diagnose bacterial CNS infection, however there is the potential for false-positive results (Nghiem and Schatzberg, 2010). Universal or consensus bacterial PCR of the 16Sr RNA gene common to all bacteria has allowed identification of Streptococcus DNA in a dog with meningoencephalitis and negative urine, blood and CSF cultures (Messer et al., 2008). Imaging of the thorax, heart and abdomen may help identify any distant septic focus.
Treatment
Definitive treatment of CNS bacterial diseases is based on isolation of the organism and determination of its antibiotic susceptibility and on the identification and elimination of the source of infection. Pending the outcome of bacterial culture and susceptibility testing or in the absence of a positive result, the initial antibacterial therapy is based on clinical findings of concomitant infection and the most likely causative agent present. The antimicrobial medications should be broad-spectrum bactericidal, have low-level protein binding and be able to penetrate the blood-brain barrier and into the abscess (in case of cerebral abscessation) (Table 5.5).
Intravenous administration is recommended for the initial 3–5 days of antibacterial treatment. Once a positive response has been achieved, the animal can be switched to oral administration with the same antimicrobial medication or an antimicrobial medication with similar spectrum (if available). Recommendations for the duration of oral antibiotic treatment vary, and generally involve several weeks of treatment.
The administration of corticosteroids in animals with CNS bacterial infections is controversial. Potential benefits include anti-inflammatory effect, which may be particularly helpful to counteract the host’s inflammatory response following bacterial lysis induced by
Structural Epilepsy

Fig. 5.8. MRI of the brain of a 7-year 11-month-old domestic short hair with subacute onset of right-sided forebrain signs and neutrophilic pleocytosis on haematology. Transverse T2W (a), T1W (b), T1WC (c), dorsal T2W (d), T1WC (e), and right-sided paramedian T1WC (f) images show signal changes in the dorsolateral aspect of the right frontal lobe and within the right frontal sinus. The affected intracranial lesion appears well demarcated (c, e, f), hyperintense in T2W (a, d) and hypointense on T1W (b) compared to grey matter. Contrast administration (c, e, f) reveals a regular band of peripheral contrast enhancement, which seems continuous with surrounding meningeal enhancement. There is midline shift to the left. The main differential diagnoses for these MRI findings are right frontal sinusitis and right frontal intracranial abscessation. The cat’s owner did not consent to surgery and the cat fully recovered with antibiotic and anti-inflammatory treatment.

bactericidal treatment. Potential detrimental effects include effect on diagnostic test results
(e.g. CSF analysis) and clinical deterioration if administered before diagnosis, and reduced blood-brain barrier permeability and consequently antibacterial medication penetration. A short course of anti-inflammatory doses of dexamethasone (0.15 mg/kg IV every 6 h for 4 days, or 0.15 mg/kg IV once followed by
0.4 mg/ kg IV every 12 h for 2 days) started 15–20 min before the first dose of antibacterial medication is considered beneficial in people with bacterial meningitis (van de Beek, 2009).
Management of brain abscessation depends on the anatomic location, number and size of abscesses, presence of foreign-body material, as well as the stage of abscess formation and neurological status of the patient. Frequently a combination of medical and surgical treatment is required (Costanzo et al., 2011). Animals with increased intracranial pressure can be administered mannitol (0.5 to 1.5 g/kg IV as a slow intravenous bolus over 15–20 min) or hypertonic saline (4 ml/kg of 7.5% sodium chloride or 5.3 ml/kg of 3% sodium chloride IV over 2–5 min) and supportive care as described in the treatment of traumatic brain injury. Anti-epileptic treatment is performed as for other types of structural brain disorders (see introduction to this chapter and Chapters 12–24). Interactions between antimicrobial and AEMs (e.g. phenobarbital and metronidazole) have to be considered. Other adjunctive therapeutic strategies, such as glycerol, paracetamol and induction of hypothermia, are being investigated in human medicine (van de Beek et al., 2012).
Prognosis
Prognosis depends on early and appropriate treatment. Prognosis has been reported to be favourable in most dogs and cats with CNS bacterial infection secondary to extension of otitis media/interna or brain abscessation secondary to bite wounds undergoing surgery and appropriate antibiotic therapy (Sturges et al., 2006; Costanzo et al., 2011). The limited information on dogs and cats with bacterial meningoencephalomyelitis suggests a guarded to poor prognosis overall, however there is no study on a large group of animals treated appropriately for confirmed bacterial meningoencephalomyelitis. The presence of seizures has not been correlated with prognosis in people with bacterial meningitis.
Table 5.5. Bactericidal antimicrobial medications for CNS bacterial infections (Riviere and Papich, 2009; Greene and Calpin, 2012; Kent, 2012).
Antimicrobial medication Spectrum Dosage Note
Ampicillin
Metronidazole
Trimethoprimsulfonamide
Third-generation cephalosporins
(e.g. cefotaxime, ceftazidime, ceftriaxone)
Gram-positive bacteria (non-betalactamase-producing staphylococci); gram-negative infections caused by Proteus and Escherichia
Anaerobic bacteria
Gram-positive and gram-negative aerobes and some anaerobes
Gram-negative bacteria resistant to other cephalosporins, usually effective against anaerobes but reduced activity against gram-positive cocci
22 mg/kg IV (slow) or SC every 6–8 h (dogs and cats)
10 mg/kg IV (slowly) or PO every 8 h
30 mg/kg every 12 h for 5 to 7 days and then 15 mg/kg every 12 h IV or PO (dogs and cats)
Cefotaxime sodium 6–40 mg/kg IV (slowly) or SC every 4–6 h (dogs and cats)
Ceftazidime 30 mg/kg IV (slowly) or SC every 4–8 h (dogs), every 8 h (cats)
Unlike other penicillins, it can reach adequate CSF concentration regardless of the state of the meninges. Alters normal intestinal microflora causing diarrhoea
Good penetration into the CSF and brain abscess. Can be combined with ampicillin. PB can increase metronidazole metabolic clearance. Can darken urine colour. Use with caution in animals with hepatic or renal
insufficiency. See Metronidazole toxicity in Chapter 4 Good penetration into the CNS. Avoid use in dogs with reduced Shirmer test, congenital
bleeding disorders, and in animals with hepatic parenchymal disease, anaemia or leukopaenia. Avoid or reduce dose in animals with renal dysfunction. Can cause immune-mediated disorders including meningitis, polyarthritis and retinitis; ataxia; salivation, diarrhoea and vomiting
Not absorbed from the gastrointestinal tract. Parenteral dosing interval shortens with increasing severity of infection. Cefotaxime is also effective against gram-positive (including beta-lactamase-producing bacteria). Ceftazidime is the most effective cephalosporin against
Pseudomonas spp. Alter normal intestinal microflora causing diarrhoea. The dosage should be reduced in patients with
renal failure
Continued
Structural Epilepsy
| Antimicrobial | |||
| medication | Spectrum | Dosage | Note |
| Carbapenems (e.g. imipenemcilastatin, meropenem) | Most gram-positive (including beta-lactamase-producing bacteria) and gram-negative aerobes and anaerobes | Imipenem-cilastatin 5–10 mg/kg IV, IM or SC every 4–8 h Meropenem 40 mg/kg IV or SC every 8 h | Poorly absorbed from the gastrointestinal tract. Can lead to development of bacterial resistance and therefore use should be avoided or limited to selected patients for treatment of microorganisms resistant to other antibacterials and for mixed infectious requiring a broad spectrum including anaerobes. Can cause seizures in patients with underlying brain disease or if overdosed. The dosage should be reduced in patients with renal failure. |
| Enrofloxacin Ciprofloxacin Marbofloxacin | Greater activity against gram-negative (especially Enterobacteriaceae) than gram-positive bacteria. Not effective against anaerobes and Enterococcus Same as for Enrofloxacin Same as for Enrofloxacin | 5–20 mg/kg PO, SC, IV every 12–24 h (dogs) 5 mg/kg every 24 h PO for 7–14 days (cats) 10 mg/kg IV every 24 h for 7–14 days 20 mg/kg PO every 24 h for 7–14 days (dogs and cats) 2.75–5.5 mg/kg PO every 24 h for 10–28 days (dogs and cats) | Renal, hepatic and hematopoietic system should be monitored during treatment May not reach therapeutic concentration in the CNS. Should not be used in growing dogs and cats as it can cause cartilage damage. Neurotoxicity (including seizures) at supratherapeutic IV doses or with rapid IV infusion or sometimes at therapeutic doses in dogs. Irreversible retinal toxicity in cats administered >5 mg/kg/day Similar to Enrofloxacin Similar to Enrofloxacin |
130 L. De Risio
Table 5.6. Ehrlichial, anaplasmal, rickettsial and mycoplasmal diseases of the CNS in dogs (Ilha et al., 2010; Sykes et al., 2010a; Headley et al., 2011; Cocayne and Cohn, 2012; Diniz and Breitschwerdt, 2012; Greene et al., 2012; Harrus et al., 2012).
Geographic Source of Haematological Disease Organism distribution infection Clinical signs abnormalities CSF analysisa Diagnosis Treatment
| Canine | Ehrlichia canis | Worldwide, | Bite of infected | Lethargy, anorexia, | |
|---|---|---|---|---|---|
| monocytotropic | tropical | tick | weight loss, fever, | ||
| ehrlichiosis | and | (Rhipicephalus | petechiae, | ||
| temperate | sanguineus) | ecchymoses, | |||
| regions, | epistaxis, | ||||
| except | lymphadenomegaly, | ||||
| Australia | splenomegaly, ocular | ||||
| signs. CNS signs, | |||||
| including seizures, | |||||
| result from vasculitis | |||||
| and/or haemorrhage, | |||||
| often have acute | |||||
| onset and may be | |||||
| focal or multifocal. | |||||
| PNS signs can also | |||||
| occur | |||||
| Canine granulo- | Ehrlichia | Mid-western | Bite of infected | Fever, lameness, | |
| cytotropic | ewingii | and | tick | stiffness, joint | |
| ehrlichiosis | southern | (Amblyomma | effusion, lethargy, | ||
| USA | americanum) | anorexia, | |||
| CNS signs | |||||
Thrombocytopaenia (moderate to severe) or pancytopaenia
Thrombo-cytopaenia; occasionally intracytoplasmic ehrlichial morulae
Increased protein concentration and mono-nuclear or mixed pleocytosis. Sometimes normal
Rarely, intracytoplasmic ehrlichial morulae within CSF monocytes
Increased protein concentration and neutrophilic or mixed pleocytosis
Rarely, intracytoplasmic ehrlichial morulae within CSF neutrophil
Measurement of specific antibodies in serum and CSF, PCR, organism detection or cultivation
Serologic testing, PCR
Doxycycline, 5–10 mg/kg every 12–24 h IV or PO for 21–28 days.
Prednisolone 1–2 mg/kg/ day for 2–7 days.
Supportive therapy
Doxycycline, 5–10 mg/kg every 12–24 h IV or PO for 14–28 days. Supportive therapy
Continued
Structural Epilepsy
Table 5.6. Continued.
Geographic Source of Haematological Disease Organism distribution infection Clinical signs abnormalities CSF analysisa Diagnosis Treatment
Canine granulo-Anaplasma cytotropic phagocytoanaplasmosis philum
Rocky Mountain Rickettsia spotted fever rickettsii
Worldwide in the temperate regions of the northern hemisphere
North, central and South America
Bite of infected tick (Ixodes spp.)
Bite of infected tick (Dermacentor, Rhipicephalus, Amblyomma)
Fever, anorexia, lethargy, lymphadenomegaly, splenomegaly, lameness, stiffness, joint swelling. Uncommonly CNS signs and haemorrhagic tendencies
Fever, lethargy, anorexia, vomiting, diarrhoea, weight loss, petechiae, ecchymoses, subcutaneous oedema, lymphadenomegaly, hepatomegaly, myalgia, arthralgia, joint swelling, conjunctivitis, anterior uveitis and retinal disease. CNS signs mainly result from vasculitis and/or haemorrhage, may be focal or multifocal, and include cervical hyperalgesia, vestibular signs, ataxia, tetraparesis, and seizures
Thrombo-cytopaenia (mild to severe) or pancytopaenia
Thrombo-cytopaenia, leukopaenia (initially), neutrophilic leucocytosis, anaemia
Increased protein concentration and neutrophilic pleocytosis
Increased protein and mononuclear, neutrophilic or mixed pleocytosis; sometimes normal
Intracytoplasmic morulae within neutrophils, seroconversion (four-fold), PCR, organism cultivation
Seroconversion (IgM or four- fold IgG); direct fluorescent antibody staining on skin lesion biopsy, PCR, organism isolation
Doxycycline, 5–10 mg/kg every 12–24 h IV or PO for 10–21 days
Supportive therapy
Doxycycline, 5–10 mg/kg every 12–24 h IV or PO for 7 days
Supportive therapy
132 L. De Risio
| Salmon | Neorickettsia | Endemic in | Ingestion of | Fever, lethargy, serous Thrombo- | Not reported | Intra- | Doxycycline, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| poisoning | helminthoeca | regions of | uncooked | to purulent ocular | cytopaenia, | cytoplasmic | 10 mg/kg | ||||||||||
| disease | the Pacific | freshwater | discharge, vomiting, | lymphopaenia, | organisms | every 12 h | |||||||||||
| Northwest | fish (most | anorexia, weight | eosinophilia, | within | IV or PO for | ||||||||||||
| of the USA | commonly of | loss, diarrhoea | frequently | macrophages | 1–2 weeks. | ||||||||||||
| and | the salmonid | (often haemorrhagic), | with increased | in lymph node Supportive | |||||||||||||
| Canada. | family) | dehydration, | band | fine needle | therapy. | ||||||||||||
| Reported | infected with | polydipsia, | neutrophils, | aspirate, | Praziquantel | ||||||||||||
| in Brazil | metacercariae | lymphadenomegaly | anaemia | isolation of | (for N. | ||||||||||||
| of the intestinal | and occasionally | the organism; | salmincola) | ||||||||||||||
| trematode | cervical | operculated | |||||||||||||||
| Nanophyetus | hyperalgesia, | trematode | |||||||||||||||
| salmincola | seizures and | eggs in the | |||||||||||||||
| containing | myoclonus | faeces of dogs | |||||||||||||||
| N. helminthoeca | 5–8 days after | ||||||||||||||||
| infection | |||||||||||||||||
| Mycoplasmosis | Mycoplasma | Worldwide | Haematogenous | Neurologic signs are | Not reported | Not reported | Identification | None proven |
| edwardii | colonization | acute in onset and | of the | |||||
| of the CNS | characterized mainly | mycoplasma | ||||||
| or direct | by seizures | (by culture or | ||||||
| inoculation. | PCR) in the | |||||||
| In the CNS | CNS at | |||||||
| secondary to | necropsy | |||||||
| trauma or | ||||||||
| surgical | ||||||||
| procedures | ||||||||
aBecause of the risk of haemorrhage, CSF collection may not be advisable in dogs with suspected rickettsial disease
Structural Epilepsy
Table 5.7. Protozoal diseases of the CNS in dogs and cats (Garosi et al., 2010; Dubey and Lappin, 2012).
Affected Source and Brain MRI CSF analysis Disease Organism species mode of infection Clinical signs findings results Diagnosis Treatment
Toxo-Toxoplasma Dog and cat. plasmosis gondii The cat is both a definitive and intermediate host
Infection can occur transplacentally, by ingestion of intermediate host tissue containing tachyzoites or bradyzoites, or by ingestion of sporulated oocystscontaminated food or water
CNS (including seizures) and/or neuromuscular signs may occur alone or in association with signs of involvement of organs
(e.g. ocular respiratory, gastrointestinal signs)
Multifocal, indistinct, contrast-enhancing parenchymal lesions, which are iso- to hypointense on T1-W images, hyperintense on T2-W images, and are associated with peri-lesional oedema
Mild to marked mononuclear or mixed pleocytosis and increased protein concentration. Occasionally normal
Quantification of IgM and IgG in serum and CSF, seroconversion, antibody coefficient >1; PCR on CSF; demonstration of T. gondii tachyzoites or bradyzoites, antibodies or antigen in tissue biopsy
Clindamycin 15 mg/ kg PO, IV, IM, SC every 12 h (dogs and cats) for 4 or more weeks
OR
Trimethoprimsulfonamide 15 mg/kg every 12 h IV, PO (dogs and cats) for 2–4 weeks
OR
Sulfonamide 20–30 mg/kg PO every 24 h and Pyrimethamine 0.25–0.5 mg/kg every 12 h for 2–4 weeks with folic acid supplementation 5 mg/day.
Supportive therapy
134 L. De Risio
| Neosporosis | Neospora caninum | Dog | The dog is both definitive and intermediate host. CNS signs | (commonly in adult dogs) | Bilaterally and symmetrically atrophied | Mild to marked mononuclear (occasionally | Quantification of IgG in serum (>1 : 800) and | Same as above |
| Infection can occur transplacentally, or by ingestion of | predominantly associated with cerebellitis, | cerebellum, surrounded by a prominent | eosinophilic) pleocytosis and moderate | CSF, PCR on CSF, demonstration | ||||
| intermediate host tissue containing tachyzoites or | neuromuscular signs (commonly in puppies) | layer, which is hyperintense on T2WI and | to marked increase in protein | of N. caninum tachyzoites or bradyzoites, | ||||
| bradyzoites | associated with polyradiculoneuritis and | hypointense on T1WI; loss of contrast | concentration. Occasionally normal | antibodies or antigen in tissue biopsy | ||||
| myositis; alone or in association | between cerebellar | |||||||
| with signs of | grey and white | |||||||
| ocular, hepatic, pulmonary, or myocardial | matter (Fig. 5.9) | |||||||
| involvement |
Structural Epilepsy


Fig. 5.9. MRI of the brain of a 7-year 6-month-old, female spayed greyhound with progressive cerebellar ataxia. Sagittal T2W (a) and transverse T2W (b), FLAIR (c), T1W (d) images at the level of the fourth ventricle. The cerebellum is bilaterally and symmetrically atrophied and surrounded by a prominent layer, which is hyperintense on T2W (b), partly attenuates on FLAIR (c) and is hypointense on T1W (d) images. There are areas of hyperintensity within the cerebellar parenchyma and poor demarcation between white and grey matter (b and c). Routine CSF analysis revealed a marked mixed pleocytosis (451 WBC/µl) and markedly increased total protein concentration (1.40 g/l). CSF PCR and serology were positive for Neospora caninum.
Table 5.8. Fungal and algal diseases of the CNS in dogs and cats (Añor et al., 2001; Salvadori et al., 2008; Lipitz et al., 2010; Sykes et al., 2010b; Bentley et al., 2011; Hecht et al., 2011; Robson and Smith, 2011; Bromel and Greene, 2012; Day, 2012a, b; Day and Barrs, 2012; Day et al., 2012; Garcia et al., 2012; Greene, R.T., 2012; Lane et al., 2012; Legendre, 2012; Márquez et al., 2012; Pressler, 2012; Sykes and Malik, 2012;Young et al., 2012).
Disease Organism Geographic distribution Mode of infection Clinical signs Diagnosis
Cryptococcosis Cryptococcus Worldwide neoformans, (Cryptococcus Cryptococcus neoformans) gattii, other Tropical and Cryptococcus spp. subtropical regions
(Cryptococcus gattii)
Inhalation of spores or yeast cells present in the environment (especially when contaminated by pigeon guano containing
C. neoformans)
Rhino-sinusitis (more common in cats than in dogs) resulting in sneezing, unior bilateral nasal discharge, epistaxis, granulomatous lesions in the nostrils, facial deformity, cutaneous lesions (more common in cats than in dogs), lethargy, inappetence, weight loss, regional or generalized lymphoadenomegaly, optic neuritis, chorioretinitis, retinal changes, rarely lameness due to osteomyelitis.
Disseminated systemic disease is common in dogs and the genito-urinary and gastrointestinal tracts may also be affected.
CNS signs occur commonly and are generally multifocal, although focal signs can also occur. Forebrain (including seizures) and/or brainstem signs are common
Microscopic identification of the organism in the CSF (with new methylene blue or India ink staining), other body fluids (e.g. nasal discharge), or infected tissue (fine needle aspirates from lymph nodes or cutaneous nodules); detection of cryptococcal polysaccharide capsular antigen by latex agglutination test on serum and/or CSF; PCR.
Culture from body fluid or infected tissue specimens and fungal susceptibility testing
Continued
Structural Epilepsy
Table 5.8. Continued.
Disease Organism Geographic distribution Mode of infection Clinical signs Diagnosis
Blastomycosis Blastomyces dermatitidis
Histoplasmosis Histoplasma capsulatum
Primarily in eastern and mid-western USA and Canada; also identified in central America, India, Africa and Europe
Temperate and subtropical regions worldwide
Inhalation of spores present in the environment. Rarely, inoculation of
B. dermatitidis into a wound
Inhalation of microconidia present in soil contaminated by bird or bat faeces
CNS signs such as obtundation, seizures and other neurological signs reflecting focal, multifocal or diffuse CNS involvement can occur without or, more commonly, with signs related to disseminated disease including anorexia, weight loss, lethargy, fever, lymphoadenomegaly, cough, dyspnoea, ocular signs (uveitis, chorioretinitis, retinal
138 L. De Risio
changes), skin lesions and lameness secondary to bone involvement
Signs related to disseminated disease are common and include anorexia, weight loss, lethargy, fever, dyspnoea, coughing, abnormal lung sounds, lymphoadenomegaly, splenomegaly, hepatomegaly, nodular or ulcerated skin lesions, ocular signs and rarely lameness secondary to bone involvement. In dogs, GI signs are common and include tenesmus and diarrhoea with mucus and fresh blood.
Neurologic involvement occurs rarely
Microscopic identification of
the organism in the CSF, other body fluids (including drainage from skin lesions, transtracheal aspirations, urine) or infected tissue (including lymph node aspirates), PCR, serology
Microscopic identification of the organism in the CSF, other body fluids or infected tissue, PCR Coccidioidomycosis Coccidioides spp.
Phaeohyphomycosis Cladophialophora bantiana (Cladosporium bantianum) and other Cladophialophora spp. Phoma eupyrena (cat)
Aspergillosis Aspergillus spp. (Fig. 5.10, Plates 5 and 6)
South-western USA, Mexico, Central and South America
Worldwide, particularly in tropical and subtropical regions
Worldwide
Inhalation of arthroconidia present in contaminated soil.
Rarely, inoculation of Coccidioides into a wound
Inhalation of spores present in the environment (soil and decomposing plants)
Inhalation of spores
Cough, fever, inappetence or anorexia, weight loss, lethargy, lymphoadenomegaly, draining skin lesions (most frequent clinical sign in cats), lameness or spinal hyperalgesia secondary to bone involvement, uveitis, keratitis, acute blindness, cardiac dysfunction, cranial and/or spinal hyperestesia, behavioural changes, ataxia, and seizures
Neurologic signs commonly reflect focal (granuloma) CNS involvement and may occur alone or with signs of systemic involvement.
Singular or multifocal ulcerating or fistulating cutaneous nodules may occur in cats
Seizures and other forebrain signs may rarely occur by extension of nasal or frontal sinus infection into the forebrain, and with disseminated aspergillosis
Microscopic identification of the organism in the CSF, other body fluids or infected tissue (by cytology, histology, immunofluorescence), measurement of specific antibodies in serum, culture
Microscopic identification of the organism in the CSF, other body fluids or infected tissue culture
Microscopic identification of the organism in body fluids or infected tissue. Serum and urine Aspergillus galactomannan antigen ELISA assay (systemic aspergillosis)
Continued
Structural Epilepsy
| Disease | Organism | Geographic distribution | Mode of infection | Clinical signs | Diagnosis |
| Protothecosisa | Prototheca zopfii and P. wickerhamii (algae) | Worldwide, infections are more common in warm, humid regions | Ingestion of contaminated material. Prototheca spp. can be isolated in raw and treated sewage, slime-flux of trees, and human and animal waste | Dogs are generally affected by the diffuse form characterized by colitis (typically haemorrhagic), multifocal neurologic signs (seizures, altered mentation, blindness, central vestibular signs), ocular signs, urinary signs and cutaneous lesions. Cats tend to | Microscopic identification of the organism on CSF and rectal mucosal scraping cytology or on CNS or other tissue histology. Culture (CSF, other body fluids or tissue) PCR |
| have the cutaneous | |||||
| form |
140 L. De Risio
aDefinitive, long-term successful treatment of CNS protothecosis has not been reported. Temporary improvement has been achieved with intrathecal and IV Amphotericin B and oral Itraconazole (Young, 2012).
| Spectrum for CNS fungal | |||
| Antifungal medication | infections | Dosage | Note |
| Amphotericin B | Cryptococcus, Blastomyces, | 0.25–0.5 mg/kg IV or 0.5–0.8 mg/kg | Poor CNS penetration, but reportedly clinically |
| deoxycholate (AMD) | Histoplasma, Coccidioides, | SC every 48 h (dogs and cats) | effective in combination with flucytosine. |
| Aspergillus spp. | Until negative serum antigen titre for | Infusion-related anaphylactic reactions can occur. | |
| Cryptococcus | Can cause nephrotoxicity. | ||
| Can cause hypokalaemia. | |||
| Heat pretreatment of AMB-d immediately before | |||
| administration decreases nephrotoxicity | |||
| Amphotericin B (AMB) | Cryptococcus, Blastomyces, | L-AMB | L-AMB is the AMB formulation that reaches the |
| encapsulated in | Histoplasma, Coccidioides, | Initial testing dose 0.5 mg/kg IV | greatest CSF concentrations. |
| unilamellar liposomes | Aspergillus spp. | Usual dose 1–3 mg/kg (dog), 1–1.5 mg/kg | Infusion-related anaphylactic reactions can occur. |
| (L-AMB) | May be used also for | (cat) IV every 48 h for 4 weeks | Can cause nephrotoxicity, although less |
| Phaeohyphomycosis | Until negative serum antigen titre for | nephrotoxic than AMD | |
| Cryptococcus | |||
| Flucytosine | Cryptococcus | 50 to 75 mg/kg PO every 8 h for | CSF concentration is 70–90% of that in serum, |
| May be used also for | 1–12 months (dogs) | good blood-brain-barrier penetration. | |
| Phaeohyphomycosis | 50 mg/kg PO every 8 h for 1–9 months (cats) or for cats ≥3.5kg 250 mg/cat | Always used in combination with AMB due to synergistic action. | |
| PO every 8 h for 1–9 months | Can cause myelosuppression, vomiting, diarrhoea, | ||
| Until negative serum antigen titre for | cutanous reaction, hepatotoxicity, renal failure, | ||
| Cryptococcus | and CNS toxicity. | ||
| Monitor haematology, renal and hepatic function | |||
| twice weekly during treatment | |||
| Fluconazole | Cryptococcus, Blastomyces, | 5–15 mg/kg IV or PO every 12–24 h | CSF concentration is 50–90% of that in plasma |
| Histoplasma, Coccidioides | for 2 or more months beyond the resolution | and independent of meningeal inflammation. | |
| May be used also for | of all signs or negative serum antigen | Can cause vomiting, diarrhoea, hepatotoxicity. | |
| Phaeohyphomycosis | titre for Cryptococcus (dogs and cats) | It can inhibit cytochrome P450 enzymes | |
| Voriconazole (VCZ) | Cryptococcus, Blastomyces, | 5 mg/kg PO or IV every 12 h (in dogs) | Good penetration in the CNS. |
| (synthetic derivative | Histoplasma, Aspergillus | 5 mg/kg PO every 12–24 h (in cats) | Retinotoxicity in dogs following IV administration. |
| of Fluconazole, broad | spp., Coccidioides | Until negative serum antigen titre for | Neurologic signs in cats administered 10 mg/kg/day. |
| spectrum triazole) | May be used also for | Cryptococcus | Potent inhibitor of cytochrome P450 enzymes. |
| Phaeohyphomycosis | Phenobarbital increases VCZ metabolic clearance |
aTreatment of CNS fungal infections also involves surgical removal or debulking of large intracranial or spinal fungal granulomas and supportive therapy. Anti-epileptic treatment is performed as for other types of structural brain disorders (see introduction to this chapter and Chapters 12–24). The choice of AEM type and dose is affected by pharmacokinetic interactions between AEMs and antifungal chemotherapy, as well as the potential hepato- or nephrotoxicity of certain antifungal medications. A short course (1–2 days) of prednisolone or dexametasone at anti-inflammatory dose can be used in case of initial neurological deterioration following AMB treatment.
Structural Epilepsy
141
Table 5.10. Parasitic diseases of the CNS in dogs and cats (Cooley, 1987 et al.; Rudmann et al., 1996; Glass et al., 1998; Windsor et al., 2009; James and Poma, 2010; Jull et al., 2012).
Geographic Disease Parasitic organism distribution Clinical signs Diagnosis Treatment
Dirofilariasis
Larva migrans
Cuterebrosis (feline ischaemic encephalopathy, meningoencephalitis in dogs)
Dirofilaria immitis microfilaria or aberrant adult migration
Aberrant larval migration of Toxocara canis, Toxocara cati, Baylisascaris procyonis, other ascaride species, or of Tenia serialis (cerebral coenurosis)
Aberrant larval migration of Cuterebra spp.
Worldwide
Worldwide (Toxocara spp., Tenia spp.);
North America (Baylisascaris procyonis)
North, central and South America
CNS signs relate to location of lesion and may be focal or multifocal
CNS signs relate to location of lesion and may be focal or multifocal
CNS signs relate to location of lesion. Peracute or acute onset lateralized or multifocal forebrain signs and seizures in cats with feline ischaemic encephalopathy
Identification of the causative parasite in the CNS at necropsy.
Presumptive diagnosis based on presence of heartworm disease and CSF pleocytosis (often including eosinophils)
Identification of the causative parasite in the CNS at necropsy.
Presumptive diagnosis based on peripheral blood eosinophilia, MRI findings: haemorrhagic tracts on T2* gradient echo sequences (Figs 5.11, Plates 7 and 8) with ascaride larval migration, large cystic lesions with Tenia serialis, CSF eosinophilic pleocytosis
Identification of the causative parasite in the CNS at necropsy.
Presumptive diagnosis based on history of upper respiratory signs 1 to 2 weeks prior to onset of intracranial neurologic signs, identification of Cuterebra larvae in the upper respiratory tract, linear regions of hypointensity on T1-weighted images, hyperintensity on T2-weighted images, and contrast enhancement, haemorrhagic tracts on T2* gradient echo sequences and CSF eosinophilic or mixed pleocytosis None proven.
Antiparasitic, antiinflammatories, antistaminics, antibiotics.
Surgical removal
None proven.
Antiparasitic, antiinflammatories, antistaminics, antibiotics.
Surgery for cerebral coenurosis
Ivermectin, antiinflammatories, antistaminics, antibiotics and supportive treatment
142 L. De Risio
Table 5.11. CNS inflammatory diseases of unknown aetiology in dogs (Higginbotham et al., 2007; Talarico and Schatzberg, 2010; Tipold and Stein, 2010).
Disease Prevalence Clinical signs Diagnosis Treatment
Steroid-responsive meningitis-arteritis (SRMA)
Granulomatous meningoencephalomyelitis (GME) (see text for more information)
Young adults, medium to large breed dogs. Breed predisposition described for Bernese mountain dogs, boxers, beagles, and Nova Scotia duck tolling retriever
Young to middle-aged, female, toy and terrier breed dogs
Severe cervical (and sometimes also thoracolumbar) hyperalgesia from inflammation of meninges and arteries. Fever. Sometimes associated with immune-mediated polyarthritis. Clinical signs are episodic and recurrent. Proprioceptive and motor deficits can occur with involvement of the neuroparenchyma (e.g. due to severe necrotizing vasculitis, infarction, or spontaneous bleeding in the subarachnoid space)
Clinical signs can be focal or multifocal depending on lesion distribution in the CNS and may include abnormal mentation, seizures, visual deficits, vestibule-cerebellar signs, paresis and cervical hyperalgesia. See Table 5.12
Presumptive diagnosis based on neutrophilic leucocytosis on haematology, severe neutrophilic pleocytosis and increased protein concentration in CSF. A combined elevation of IgA levels in serum and CSF supports the diagnosis with a high sensitivity, but low specificity. Acute phase proteins (APPs), including C-reactive protein (CRP), are elevated in the serum and CSF. MRI can show meningeal enhancement and enlarged vessels in severe cases.
Definitive diagnosis by histology of the meninges and neuroparenchyma
Presumptive diagnosis based on clinical presentation, MRI and CSF findings (see text) and negative infectious disease testing.
Definitive diagnosis by histology of the brain (biopsy or post-mortem)
Immunosuppressive doses of prednisone, tapered off gradually over up to 6 months: 2 mg/kg body weight every 12 h for 1 to 2 days, then reduced to 1 mg/kg every 12 h for 1 to 2 weeks, than slowly reduced based on clinical response and CSF analysis, haematology and serum CRP levels until a dose of 0.5 mg/kg every other day is reached. Additional immunosuppressive medications can be used in refractory cases
Immunosuppressive doses of prednisone alone or often in combination with one or more of the following medications: cytosine arabinoside, cyclosporine, procarbazine, mycophenolate mofetil, lomustine, leflunomide
Continued
Structural Epilepsy
| Disease | Prevalence | Clinical signs | Diagnosis | Treatment |
| Necrotizing meningoencephalitis (NME) Necrotizing leukoencephalitis (NLE) | Pug, Maltese terrier, shihtzu, French bulldog, Lhasa apso, Chihuahua, Pekingese, West Highland white terrier and other small dog breeds Yorkshire terrier, French bulldog, Maltese terrier and other small dog breeds | Neurologic signs reflect involvement of the forebrain and include altered mentation, focal or generalized seizures, visual deficits, circling, head pressing and sometimes cervical hyperalgesia Neurologic signs reflect involvement of both forebrain and brainstem and include altered mentation, visual deficits, seizures, proprioceptive deficits and central vestibular signs | Presumptive diagnosis based on clinical presentation, MRI (asymmetric, multifocal forebrain lesions with signal changes similar to GME, loss of cerebral grey/white matter distinction) and CSF (lymphocytic pleocytosis and elevated protein concentration) findings, and negative infectious disease testing. Definitive diagnosis by histology of the brain Presumptive diagnosis based on clinical presentation, MRI (multiple, asymmetric bilateral lesions with signal changes similar to GME and multiple areas of necrosis affecting the white matter of the forebrain, mainly in the corona radiata, centrum | See GME See GME |
| semiovale and internal | ||||
| capsule, and brainstem) and CSF (moderate mononuclear pleocytosis and elevated protein concentration) findings, and negative infectious disease testing. Definitive diagnosis by histology of the brain |
144 L. De Risio
Structural Epilepsy

Fig. 5.10. Transverse T2-weighted MRI at the level of the lateral ventricles of a 5-year-old female vizsla which presented with seizures and circling. There is an asymmetrical mass lesion affecting the entire right-sided cerebral hemisphere, extending into the thalamus and brainstem. Extensive hyperintensity within the white matter of the cerebrum suggests secondary oedema.
Granulomatous meningoencephalomyelitis
Granulomatous meningoencephalomyelitis (GME) is an inflammatory disorder of the brain whose aetiopathogenesis remains unknown. Autoimmune, infectious, neoplastic, genetic and even toxic aetiologies have been proposed (Talarico, 2010). Most likely GME is a nonspecific immunologic response associated with multiple environmental triggers (including pathogens and vaccinations) as well as genetic factors (Talarico, 2010). Typically, GME presents as an acute-onset, progressive, neurologic disease that may be fatal if left untreated. Young to middle-aged dogs, females and toy and terrier breeds are over-represented; however, dogs of any age, gender and breed may be affected (Talarico and Schatzberg, 2010).
Clinical signs
Clinical manifestations and lesion topography of GME can vary. Three basic forms of the disease have been reported: ocular, focal and disseminated or generalized (Table 5.12). The disseminated form is the most common (Talarico and Schatzberg, 2010). The focal form of GME should be differentiated from CNS malignant histiocytosis and primary CNS lymphoma.
Diagnostic investigations
Haematology, serum biochemistry and urinalysis may be normal or reveal aspecific changes. Sensitivity and specificity of high-field routine MRI in the diagnosis of GME is 50% and 88%, respectively (Wolff et al., 2012). MRI features of GME include lesions that are typically hyperintense on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences, iso- to hypointense on T1-weighted images and variably contrast-enhancing, ranging from none to intense contrast uptake. In the disseminated form of GME these lesions typically have an infiltrative appearance with irregular margins and are multifocal (Fig. 5.12 a–e). Although GME has a predilection for white matter, it is not associated with distinct topography on MRI, as are NME and NLE, and MRI lesions often are distributed throughout both grey and white matter (Higginbotham et al., 2007; Talarico and Schatzberg, 2010). A focal space-occupying mass or abnormalities involving the optic nerves and chiasm may be observed in animals with the focal or ocular forms, respectively. CSF analysis can reveal mild to severe mononuclear (and less commonly, mixed) pleocytosis and elevation of total protein concentration; however CSF can occasionally be normal. As the MRI and CSF findings are not specific for GME, the ante-mortem diagnosis of this disease is also based on exclusion of infectious or neoplastic CNS disorders by means of various tests including serology, CSF PCR, thoracic and abdominal imaging. Definitive diagnosis requires histological evaluation of the brain through biopsy or post-mortem. Histologically, GME is characterized by large perivascular cuffs of mononuclear cells sometimes distributed in a whorled pattern,

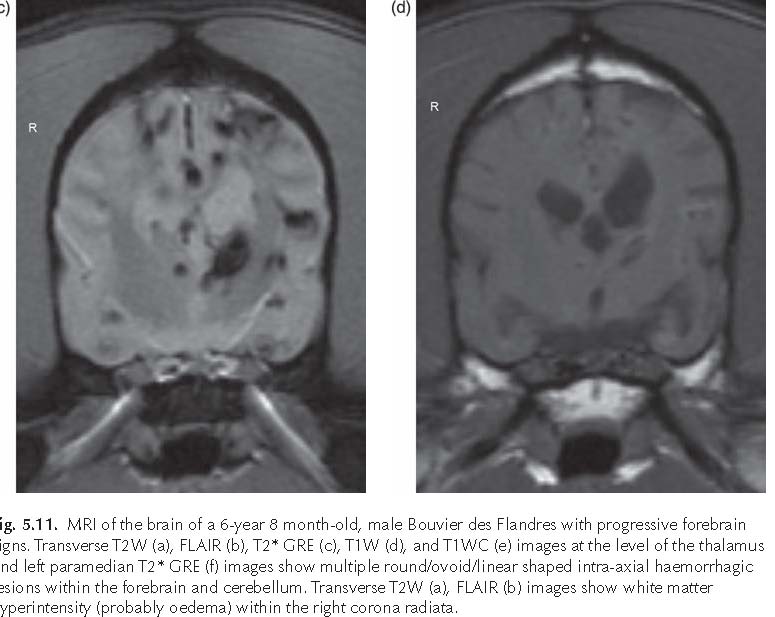
Structural Epilepsy
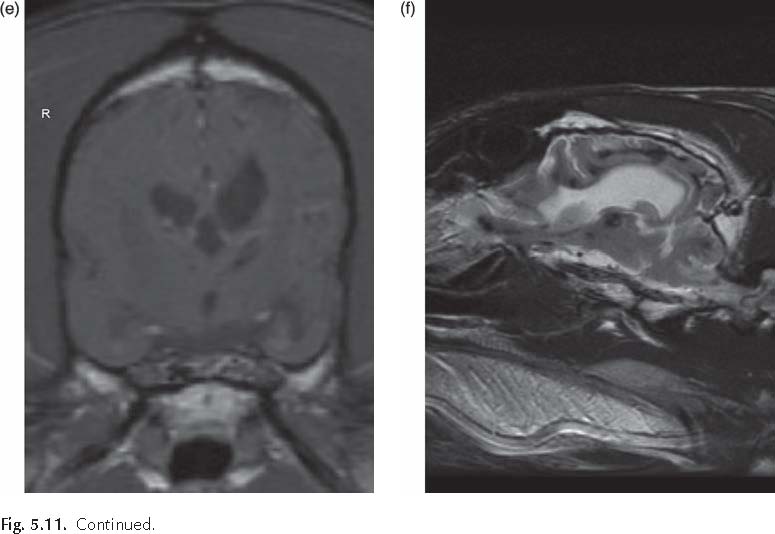
within the neuroparenchyma and meninges (Plate 9). Unlike NME and NLE, tissue necrosis and secondary cavitation are lacking (Higginbotham et al., 2007).
Treatment
Immunosuppression is the mainstay of therapy for GME as well as other MUE (meningoencephalitis of unknown aetiology). Gold-standard treatment protocols have yet to be established due to the lack of prospective randomized and blinded clinical trials comparing different standardized treatment protocols in dogs with GME or MUE (Granger et al., 2010). In clinical practice prednisone or dexamethasone treatment is commonly initiated when GME is suspected based on clinical and diagnostic investigation findings. The dosage of corticosteroid and whether and when additional immunosuppressive agents are started depend on the clinician preference, index of suspicion of GME (or other MUE) and the severity of neurological dysfunction. Anti-inflammatory dosage of corticosteroids (e.g. prednisolone
0.5 mg/kg to 1.0 mg/kg once daily) and antibiotic therapy are often administered while awaiting serology and PCR results for regional infectious diseases. However, if the index of suspicion of GME (or other MUE) is high and neurological dysfunction is severe, immunosuppressive dosage of prednisolone (1 to 2 mg/kg every 12 h) alone or in combination with another immunosuppressive medication may be started immediately. Prednisolone protocol can vary depending on clinician preference, severity of neurological dysfunction, response to treatment, development of steroid-related adverse effects, pet-owner financial and personal situation and availability of other immunosuppressive medications. The aim of long-term prednisolone treatment is to find the lowest dose to control neurological signs in each dog. An example of a possible prednisolone protocol is summarized in Box 5.3.
Adverse effects associated with long-term, high-dose corticosteroid therapy include polyuria–polydipsia, polyphagia, weight gain, hepatotoxicity, gastrointestinal ulceration, pancreatitis and iatrogenic hyperadrenocorticism. Therefore adjunctive immunosuppressive treatment is often preferred to minimize
Table 5.12. Features of the different forms of GME.
Form of GME Lesion location Clinical signs Disease onset and course
Ocular GME Retinal and post-retinal portions of the optic nerve and optic chiasm
Focal GME Single lesion located in the cerebrum, brainstem (especially in the pontomedullary region), or, rarely, cerebellum or spinal cord
Disseminated or Multifocal lesions generalized GME involving primarily the white matter of the cerebrum, caudal brainstem, cerebellum, and cervical spinal cord; however, grey matter, leptomeninges and choroid plexus may also be involved
Visual dysfunction, commonly dilated and unresponsive pupils, variable degrees of optic disc edaema, and occasionally chorioretinitis, especially in the nontapetal fundus
Related to the location of the single space-occupying mass lesion
Related to the location of the lesions. Commonly include altered mentation, visual deficits, seizures, vestibulocerebellar and cervical spinal cord signs
Acute onset. May occur concurrently or progress to disseminated form
Acute to chronic onset, rapidly or slowly progressive
Acute onset and rapidly progressive
the prednisolone dosage while reaching and maintaining optimal clinical outcome.
Reported adjunctive immunosuppressive medications include cytosine arabinoside, procarbazine, cyclosporine, lomustine, leflunomide and mycophenolate mofetil.
Cytosine arabinoside (CA) is a chemotherapeutic agent with immunosuppressive properties that crosses the BBB in dogs. CA is a synthetic nucleoside analogue that competitively inhibits DNA polymerase in mitotically active cells, causes topoisomerase dysfunction, prevents DNA repair, and inhibits ribonucleotide reductase and glycoprotein synthesis. CA is metabolized by deamination in the liver, plasma, granulocytes and gastrointestinal tract (Talarico and Schatzberg, 2010). CA is commonly administered at 50 mg/m2 subcutaneously twice daily for 2 consecutive days. This treatment cycle is repeated every 3 weeks for three cycles. Subsequently, the interval between treatment cycles is increased by 1 week and the dog receives three treatment cycles at the new treatment interval. After three treatments, the interval between treatment cycles is extended by another week. The new interval is maintained for three treatment cycles. Treatment cycle intervals are gradually extended. Concurrently, the dose of prednisolone is gradually tapered to a low dosage administered every other day. In severely affected dogs CA can also be administered intravenously as constant rate infusion at 200 mg/m2 over 48 h (de Stefani et al., 2008). A recent pharmacokinetic study showed that steady-state plasma concentrations were achieved after approximately 4 h when CA was administered via intravenous constant rate infusion (CRI), but did not reach steady state after subcutaneous (SC) administration due to rapid absorption and elimination (Crook et al., 2013). The steady state achieved with CA intravenous CRI may produce a more prolonged exposure of CA at cytotoxic concentrations in plasma compared to the concentrations after SC administration, improve penetration of CA across the BBB and produce higher efficacy for treatment of MUE
Structural Epilepsy


Fig. 5.12. MRI of the brain of a 6-year 3-month-old, female West Highland white terrier with chronic progressive multifocal intracranial signs. Sagittal T2W (a) and transverse T2W (b), FLAIR (c), T1W (d) and T1WC (e) images reveal diffuse poorly defined signal changes within the brainstem, which appear hyperintense on T2W (a, b) and FLAIR (c), isointense on T1W (d) and heterogeneously contrast enhancing on T1WC (e). The sagittal T2W (a) image shows similar changes within the cerebral white matter and cranial cervical spinal cord. The main differential diagnosis was GME. Neurological signs resolved following treatment with corticosteroids and cytosine arabinoside.

Box 5.3. Prednisolone protocol for GME.
2 mg/kg twice daily for 1–2 days 1 mg/kg twice daily for 4 weeks
0.5 mg/kg twice daily for 4–8 weeks
0.5 mg/kg once daily for 4–8 weeks
0.5 mg/kg every other day for 8–16 weeks or indefinitely
0.25 mg/kg every other day for 8–16 weeks or indefinitely
(Crook et al., 2013). Adverse effects of CA are dose-dependent and include myelosuppression, vomiting, diarrhoea, hair loss and calcinosis cutis following subcutaneous administration (Scott-Moncrieff et al., 1991; Volk et al., 2012). Haematology should be performed prior to each treatment course and 10 to 14 days after the first course of CA. Reported median survival time of dogs with GME or MUE treated with CA in combination with corticosteroids ranges from 26 to 735 days (Zarfoss et al., 2006; de Stefani et al., 2008; Menaut et al., 2008; Smith et al., 2009; Lowrie et al., 2013).
Procarbazine is lipid soluble antineoplastic, alkylating agent that crosses the BBB. It alkylates DNA at the O6 position of guanine, inhibiting insertion of essential DNA precursors and disrupts ribonucleic acid and protein synthesis. Procarbazine has been used as an adjunctive therapy with corticosteroids or as monotherapy for MUE at a dose of 25 to 50 mg/m2/day orally. The procarbazine dose can be reduced to every other day, if clinical improvement is observed after the first month of treatment and in the absence of relapses. Adverse effects include myelosuppression, nausea, vomiting, haemorrhagic gastroenteritis, hepatic dysfunction and neurotoxicity. Haematology should be checked once weekly for the first month of treatment and monthly thereafter. Median survival time for dogs treated with procarbazine in combination (at least initially) with corticosteroids is 425 days (Coates et al., 2007).
Cyclosporine is an immunosuppressive agent that acts by suppressing T lymphocyte activation and proliferation both directly and indirectly by preventing synthesis of several
Structural Epilepsy
cytokines including interleukin-2. Cyclosporine has been used as a monotherapy or more commonly in combination with prednisone or ketoconazole in dogs with MUE. The dosage as monotherapy is 6 mg/kg orally every 12 h (Adamo et al., 2007a, b). The dosage of cyclosporine is 5 mg/kg orally once a day when used in combination with ketoconazole at 8 mg/kg orally once a day (Adamo et al., 2007a, b). Ketoconazole lowers the dose of cyclosporine needed to achieve effective blood levels by inhibiting the cytochrome P450 enzymes and decreasing cyclosporine systemic clearance. Concurrent administration of phenobarbital will decrease cyclosporine blood levels as phenobarbital induces the P450 enzyme which metabolizes cyclosporine. Therefore this combination is not ideal in seizuring dogs with GME or other MUE. The dosage of cyclosporine should be adjusted to achieve blood levels between 200 and 400 ng/ml (Adamo et al., 2007a, b). The serum cyclosporine trough level should be tested 7 days after initiation of treatment and re-evaluated after 1 month and subsequently every 4 or 6 months, or in case of neurological deterioration (Adamo et al., 2007a, b). The most common adverse effects of cyclosporine include diarrhoea, anorexia and vomiting. Occasionally, gingival hyperplasia, papillomatosis, hypertrichosis and excessive shedding may occur. Rare adverse effects include nephrotoxicity and/or hepatotoxicity. Ketoconazole adverse effects include anorexia, vomiting, diarrhoea and, rarely, hepatotoxicity. Median survival time for dogs treated with cyclosporine alone or in combination with corticosteroids and/or ketoconazole ranges from 240 to 930 days (Gnirs, 2006; Adamo et al., 2007b; Pákozdy et al., 2009).
Lomustine (CCNU) is an antineoplastic nitroso urea compound with potent immunosuppressive properties due to its toxic effect on lymphocytes. It alkylates both DNA and RNA. CCNU is highly lipid soluble and readily crosses the BBB. CCNU at 60 mg/m2 orally every 6 weeks has been used in combination with tapering doses of prednisone in dogs with MUE (Flegel et al., 2007; Uriarte et al., 2008). Adverse effects include myelosuppression, vomiting, diarrhoea and hepatotoxicity. Periodic haematology and serum biochemistry monitoring is recommended. Median survival time for dogs treated with CCNU in combination with corticosteroids is approximately 300 days (Flegel et al., 2007; Uriarte et al., 2008).
Leflunomide is an immunosuppressive medication that inhibits T- and B-cell proliferation, suppresses immunoglobulin production and interferes with cell adhesion. Leflunomide has been used at 1.5 to 4 mg/kg orally once daily in those dogs with MUE and a poor or adverse response associated with prednisolone therapy (Gregory et al., 1998). Adverse effects include thrombocytopaenia and haemorrhagic colitis. Reported median survival time for a small number of dogs with MUE treated with leflunomide in combination with corticosteroids is 365 days.
Mycophenolate mofetil is a lymphocyte-specific immunosuppressive medication that decreases the recruitment of inflammatory cells. Mycophenolate mofetil at a dose of 20 mg/kg orally twice daily for 1 month and subsequently decreased to 10 mg/kg twice daily has been used in combination with tapering doses of prednisone in dogs with GME (Feliu-Pascual et al., 2008). Adverse effects include haemorrhagic diarrhoea, myelosuppression and hepatoxicity. Reported median survival time in a pilot study including a small number of dogs treated with mycophenolate mofetil in combination with corticosteroids was 118 days (Feliu-Pascual et al., 2008).
Radiation therapy has also been reported for the treatment of focal GME in the forebrain (Munana and Luttgen, 1998). In a study including only dogs with histological diagnosis of GME, median survival was 14 days (range 1–1215 days) (Munana and Luttgen, 1998).
Antiepileptic treatment is performed as for other types of structural brain disorders (see introduction to this chapter and Chapters 12–24). Potassium bromide or zonisamide may be preferred to phenobarbital for long-term treatment of dogs receiving high doses of steroids, potentially hepatotoxic immunosuppressive agents or medications that interfere with or are metabolized by the cytochrome P450 enzymes. In addition, even though rare, the potential for phenobarbital-induced haematological abnormalities has to be considered as this may interfere with the use of potentially myelosuppressive immunosuppressive medications (e.g. cytosine arabinoside).
Prognosis
Overall, prognosis seems to improve when prednisolone is used with an adjunctive immunosuppressive medication, however there has been no prospective, randomized study to investigate this anecdotal finding. The overall reported median survival for dogs (n = 91) treated only with corticosteroids ranged from 28 to 357 days. In comparison, the overall reported median survival for dogs (n = 96) treated with corticosteroids in combination with any other immunosuppressive medication ranged from 26 to 930 days.
One study (Munana and Luttgen, 1998) suggested that dogs with focal forebrain signs survive longer than dogs with multifocal signs or focal brainstem signs. However, another study identified a significant association between the presence of seizures and reduced survival time in dogs with GME (Coates et al., 2007). A more recent study (Lowrie et al., 2013) found no associations between survival time and occurrence of seizures, age at presentation, CSF cell count or CSF protein concentration at initial diagnosis. A significant association was identified between mortality and evidence of foramen magnum herniation, loss of cerebral sulci and mass effect on MRI. Transtentorial herniation, rostral or caudal fossa involvement, and the presence of post-contrast hyperintense lesions were not associated with mortality. A normal MRI of the brain at the 3 month reexamination was significantly associated with a good or excellent outcome. An abnormal CSF analysis at the 3 month re-examination, despite a normal MRI scan, was associated with an increased risk of relapse of neurological signs when treatment was tapered (Lowrie et al., 2013).
Traumatic Brain Injury
Traumatic brain injury (TBI) in dogs and cats occurs most commonly secondary to accidental impact with motor vehicles. Other causes include gunshot or pellet injuries, animal bites, kicks and falls (Kolataet et al., 1974; Streeter et al., 2009).
Pathophysiology of TBI
Pathophysiologically, TBI can be classified into primary and secondary injury. Primary TBI refers to the physical disruption of intracranial structures at the time of the traumatic incident, which initiates a number of interrelated biochemical events that characterize the secondary injury. Primary TBI includes direct damage to brain parenchyma (such as contusion, laceration and diffuse axonal injury) and cerebral blood vessels resulting in intracranial haemorrhage (epidural, subdural, subarachnoid, intraparenchymal) and vasogenic oedema. Unstable or depressed skull fractures can perpetuate the trauma to the cerebral parenchyma and blood vessels. The extent of the primary TBI is affected by the degree of the acceleratory/deceleratory and rotational impact forces. Secondary TBI occurs in the minutes to days following the trauma and involves several interrelated and self-perpetuating events including:
The biomolecular events that characterize secondary TBI are perpetuated and exacerbated by cerebral ischaemia, which can be aggravated by systemic hypotension and hypoxaemia.
Both primary and secondary TBI contribute to increase intracranial pressure (ICP)
Structural Epilepsy
and cerebral tissue damage. ICP is the pressure exerted within the skull by the intracranial content. An increase in ICP results in a decrease in cerebral perfusion pressure (CPP) and therefore reduced cerebral blood flow, oxygenation and trophism. CPP is determined by mean arterial blood pressure (MABP) and ICP, where:
CPP = MABP – ICP (5.1)
In the normal animal due to a phenomenon called pressure autoregulation, ICP remains constant when MABP is between 50 and 150 mmHg. An increase in MABP results in cerebral vasoconstriction and a decrease in MABP results in cerebral vasodilation. Following TBI, pressure autoregulation may be lost and systemic hypotension may result in decreased CPP in the presence of an increased ICP. Cerebral ischaemia will further exacerbate the secondary injury processes leading to cerebral oedema and precipitating increased ICP. Cerebral blood flow is also chemically autoregulated as cerebral blood vessels respond directly to the arterial partial pressure of carbon dioxide (PaCO2). Elevated PaCO2 causes cerebral vasodilation, while decreased PaCO2 causes cerebral vasoconstriction. This form of autoregulation often remains intact in people with TBI.
Another compensatory mechanism, called intracranial compliance, results in a decrease in intracranial CSF and blood volume secondary to an increase in the volume of intracranial content (due to oedema and haemorrhage in patients with TBI) in order to minimize the increase in ICP. When intracranial compliance mechanisms are exhausted, a further small increase in intracranial volume will result in dramatic elevations of ICP causing severe cerebral ischaemia, rapid neurological deterioration and potentially life-threatening caudal transtentorial and/or foramen magnum cerebral herniation (Plate 10). Severe increases in ICP trigger the cerebral ischaemic response, or Cushing reflex characterized by elevation in MABP and bradycardia (Sande, 2010). In addition, an increase in ICP and resultant decrease in CPP stimulates the release of catecholamines which can lead to the brain-heart syndrome, characterized by a variety of cardiac arrhythmias resulting from myocardial ischaemia.
Initial assessment and emergency treatment
Initial clinical assessment and emergency treatment of TBI focus on the ABCs (airway, breathing, cardiovascular status) of emergency medicine and imminently life-threatening abnormalities (Sande, 2010). Hypovolaemia and hypoxaemia must be recognized and treated immediately as they affect ICP increase and cerebral damage. The initial emergency tests include packed cell volume and total protein to assess the degree of blood loss; blood glucose; blood pressure; electrocardiography; electrolytes; and arterial blood gas analysis to assess perfusion, ventilation, oxygenation and acid-base status. Once normovolaemia and appropriate oxygenation and ventilation are established (see Table 5.13), the patient is carefully examined for other traumatic injuries and a complete neurologic examination can then be performed.
A modified version of the Glasgow Coma Scale (MGCS) designed for humans has been proposed in veterinary medicine for dogs with TBI to grade the neurological status on admission and to monitor any response to treatment (Shores, 1989; Platt et al., 2001). The scale incorporates three categories of the examination (Box 5.4): level of consciousness, posture and limb motor function, and brainstem reflexes, with a score of 1 to 6 being assigned to each domain. The score in each domain is summed, yielding the total MGCS, which can range from 3 (severe neurological dysfunction) to 18 (neurologically normal). Neurological assessment should be repeated every 30 to 60 min depending on the severity of neurological dysfunction following TBI. Signs consistent with raised ICP (e.g. decreasing MGCS score, systemic hypertension with bradycardia, brain-heart syndrome) should be recognized and treated promptly and aggressively.
Post-traumatic seizures and epilepsy
Seizures can occur at any time (hours to years) after TBI. The inciting head injury is usually, but not always, severe enough to cause unconsciousness at the time of the impact. Post-traumatic seizures have been classified as immediate, early and late depending on time of occurrence (less than 24 h, 1 to 7 days
Table 5.13. Monitoring parameters, recommended goals and treatment for animals with head trauma.
Monitoring parameter Recommended goal Recommended treatment
Respiratory rate 10–25/min and rhythm
Heart rate and 70–150 bpm (dogs)
rhythm 80–180 bpm (cats) Avoid tachy- or bradycardia Avoid arrhythmias
Blood pressure MABP 80–120 mmHg Systolic BP >90 mmHg
Oxygen saturation SPO2 ≥95% of haemoglobin (pulse oximetry)
Arterial blood gasses PaO2 ≥90 mmHg
PaCO2 <35–40 mmHg Central venous pressure 5–12 cm H2O Body temperature 37–38.5°C (98.6–101.3°F)
Neurological MGCS >15 examination
ICP 5–12 mmHg Blood glucose 4–6 mmol/l (72–108 mg/dl)
Electrolytes Within laboratory normal values
Oxygen supplementation (100 ml/kg/min) Thoracocentesis in animals with pneumothorax Analgesia Consider intubation and mechanical ventilation Addressed ICP Fluid therapy (see text) Analgesia Addressed ICP Treat arrhythmias specifically Fluid therapy Vasopressor support (dopamine 2–10 µg/kg/min) Oxygen supplementation (50–100 ml/kg/min) Consider intubation and mechanical ventilation
Oxygen supplementation (50–100 ml/kg/min) Consider intubation and mechanical ventilation Fluid therapy Passive warming or cooling Paracetamol or NSAIDs if hyperthermic Elevate the head by 15–30° (see text) Consider use of hyperosmotic agents and
surgery (see text) As above for treatment of MGCS <15 Fluid therapy (consider dextrose administration,
but avoid hyperglycaemia) Fluid therapy; supplement fluids if needed
Box 5.4. Modified Glasgow Coma Scale (Platt et al., 2001).
| Level of consciousness | Score |
| Occasional periods of alertness and responsive to environment | 6 |
| Obtundation or delirium, capable of responding but response may be inappropriate | 5 |
| Obtunded, responsive to visual stimuli | 4 |
| Obtunded, responsive to auditory stimuli | 3 |
| Stuporous, responsive only to noxious stimuli | 2 |
| Comatose, unresponsive to repeated noxious stimuli | 1 |
| Posture and limb motor function | Score |
| Normal gait, normal spinal reflexes | 6 |
| Hemiparesis, tetraparesis or decerebrate activity | 5 |
| Recumbent, intermittent extensor rigidity | 4 |
| Recumbent, constant extensor rigidity | 3 |
| Recumbent, constant extensor rigidity with opisthotonus | 2 |
| Recumbent, hypotonia of muscles, depressed or absent spinal reflexes | 1 |
| Brainstem reflexes | Score |
| Normal pupillary light reflexes and vestibulo-ocular reflexes | 6 |
| Slow pupillary light reflexes and normal to reduced vestibulo-ocular reflexes | 5 |
| Bilateral unresponsive miosis with normal to reduced vestibulo-ocular reflexes | 4 |
| Pinpoint pupils with reduced to absent vestibulo-ocular reflexes | 3 |
| Unilateral, unresponsive mydriasis with reduced to absent vestibulo-ocular reflexes | 2 |
| Bilateral, unresponsive mydriasis with reduced to absent vestibulo-ocular reflexes | 1 |
Structural Epilepsy
and more than 7 days, respectively) following head trauma (Agrawal et al., 2006; Lowenstein, 2009). Recurrent late onset post-traumatic seizures are referred to as post-traumatic epilepsy (PTE). PTE presents with different seizure frequency and may evolve into remission or develop into intractable seizures (Chen et al., 2009).
The pathogenesis of post-traumatic seizures is incompletely understood and most likely multifactorial. TBI likely activates multiple pathophysiologic epileptogenic processes simultaneously or sequentially including alterations in neurotransmitter concentrations, neurotransmitter receptors, ion channels, GABA-ergic interneurons and glial cells as well as formation of excessive new excitatory synaptic connectivity (Prince et al., 2009).
The incidence of early and late post-traumatic seizures in the civilian population ranges from 2.1 to 16.3% and 1.9 and 25.3%, respectively (Lowenstein, 2009). PTE represents the cause of symptomatic epilepsy in approximately 20% of the general population (Agrawal et al., 2006). The incidence of PTE in people ranges between 1.8 and 53% depending on population studied (civilians versus soldiers), severity of head injury and follow-up duration (Frey, 2003; Diaz-Arrastia et al., 2009; Lowenstein, 2009). The risk to develop PTE increases with increased severity of head injury (Lowenstein, 2009; Ferguson et al., 2010). In a population-based study, the 5-year cumulative probability of seizures was 0.7% in patients with mild TBI (GCS 13–15), 1.2% with moderate TBI (GCS 9–12) and 10.0% with severe TBI (GCS 3–8) (Annegers et al., 1998; Hung and Chen, 2012). Evidence of damage on neuroimaging increases the risk of developing PTE by 10 to 20% (Hung and Chen, 2012). Intracranial haemorrhage increases the risk of PTE by 10-fold (Hung and Chen, 2012). Additional risk factors for developing PTE include early and late post-traumatic seizures, length of post-traumatic amnesia, loss of consciousness, coma duration, low Glasgow Coma Scale, lesion location, focal neurologic deficits, depressed skull fractures, cerebral contusions, retained bone and metal fragments, brain parenchyma loss and persistent focal electroencephalographic (EEG) abnormality.
A family history of epilepsy may suggest a higher risk of PTE in patients with mild or severe TBI (Christensen et al., 2009; Hung and Chen, 2012).
In dogs with head injury the overall incidence of post-traumatic seizures (early and late) has been reported as 5.4% (14/259) and 18.6% (44/236), respectively, in two studies (Friedenberg et al., 2012; Steinmetz et al., 2013). Both studies concluded that dogs with head trauma had a greater risk to develop epilepsy than the general canine hospital population and severity of head injury was associated with the risk to develop post-traumatic seizures. Observed seizure types include focal with or without secondary generalization and generalized tonic-clonic, isolated seizures, cluster seizures and status epilepticus (Hayes, 2009; Friedenberg et al., 2012; Steinmetz et al., 2013). The study including 259 dogs with head trauma reported early seizures in 3.5% (9/259) dogs and PTE in 5 of 74 (6.8%) dogs with long-term follow up. None of the five dogs that developed PTE had early seizures, however follow-up information was available for only one of the nine dogs with early seizures (Friedenberg et al., 2012). In the study including 236 dogs with traumatic head injury, 33 (14%) developed early seizures, predominantly within 24 h post-injury (Steinmetz et al., 2013). Thirteen (39%) of these 33 dogs died or were euthanized within the first week post-injury. Five other dogs with head injury and no seizures died within the first week after the injury. Mortality was significantly higher in dogs with early seizures after head trauma than in those with no seizures after head trauma. Of the 198 dogs that did not die within 1 week following head injury, 13 dogs (6.6%) developed late recurrent seizures (PTE). The average latency between head injury and onset of PTE was 1 year. Only 2 of the 20 dogs that survived head trauma associated with early seizures developed late seizures. Open skull injury was associated significantly more often with seizures (early or late) than closed injury. In addition, 14.3% of the dogs with open skull injury developed PTE compared to 5.3% of the dogs with closed injury. Of the 33 dogs with open skull injury, nine had penetrating injury. Of the dogs with penetrating injury, 44% (4/9) developed early seizures, whereas none developed PTE, most likely because only two dogs survived the first week after head injury. Six of nine dogs with an MRI diagnosis of intracranial haemorrhage exhibited early seizures, suggesting that such haemorrhage was a risk factor for early seizures. None of these dogs with intracranial haemorrhage developed late seizures (Steinmetz et al., 2013).
PTE has been reported in cats (see Quesnel et al., 1997; Chapter 8). However, in a recent study, none of 52 cats with mild (MGCS score 15 to 18) or moderate (MGCS score 9 to 14) TBI developed seizures during a follow-up period of 2 years or longer after head trauma (Grohmann et al., 2012).
Immediate and early post-traumatic seizures must be treated aggressively as they can result in cerebral hypoxia, oedema and increased ICP. The choice of the type and dosage of AEM depend on the patient’s neurological status and concurrent injuries to other organs (see Chapters 12, 23 and 24). In addition, the effect of AEMs on mental status and respiratory function should be carefully considered. Diazepam (0.5 mg/kg IV from once up to three times in 24 h) and phenobarbital (2–3 mg/kg IV, IM, PO as single or multiple administrations repeated up to 18–24 mg/kg over a 24–48 h period, followed by 2–3 mg/kg IV, IM, PO every 12 h) are commonly used (see Chapter 24). Levetiracetam (20–60 mg/kg IV, PO once, followed by 20 mg/kg every 8 h) may represent a safe and effective alternative or addition to DZP and PB for short-term treatment of immediate and early post-traumatic seizures (see Chapters 16 and 24). Refractory immediate and early post-traumatic seizures require additional therapy such as continuous infusion of DZP (0.1–0.5 mg/kg/h diluted in dextrose saline) or propofol (4–8 mg/kg bolus to effect followed by 1–5 mg/kg/h continuous infusion) (see Chapter 24).
Anti-epileptic treatment of PTE is performed as for other types of structural brain disorders (see Chapters 12–22). The benefits of prophylactic anti-epileptic treatment at any time after head injury have not been investigated in veterinary medicine and remain controversial in people despite randomized controlled trials. Prophylactic anti-epileptics (including levetiracetam) are effective in reducing the incidence of early seizures, but there is no evidence that prophylactic anti-epileptic treatment reduces the occurrence of late seizures or has any effect on incidence of death and neurological disability (Schierhout and Roberts, 2012). The American Academy of Neurology and the Brain Trauma Foundation both make the recommendation to initiate AEM (phenytoin or levetiracetam) in the acute setting of severe TBI to prevent provoked or early seizures. Anti-epileptic prophylactic treatment is not continued beyond 7 days after TBI. There is no evidence to support that glucocorticoids prevent the development of PTE (Hung and Chen, 2012). Further understanding of epileptogenesis following TBI would enable identifying targets for PTE prophylaxis (Huifang et al., 2011).
Diagnostic investigations
The diagnosis of TBI is often based on the history and clinical signs of intracranial dysfunction. Additional investigations to the initial emergency tests on blood and urine may be indicated. Radiography of the vertebral column, thorax and abdomen are indicated to investigate injuries to other organs. Skull radiography may reveal calvarial fractures, but provides no information on the brain parenchyma. Unless the animal is comatose, brain imaging requires general anaesthesia which can destabilize the TBI patient. Brain CT or MRI should be reserved to animals that are refractory to aggressive medical treatment, and those with progressive neurological dysfunction. Brain imaging may help to determine whether surgical treatment is indicated as in the case of a depressed skull fracture or intracranial haemorrhage (Fig. 5.13a–e).
ICP can be monitored through placement of a pressure or fibre optic transducer into the epidural, intra-axial, or intraventricular space. Risks associated with ICP monitoring include oedema, haemorrhage, parenchymal damage and infection. ICP monitoring is not frequently used in veterinary medicine due to technical and financial limitations. In human medicine ICP monitoring is considered the standard of care for severe TBI, however, there is contradicting evidence about whether ICP monitoring improves outcome. In a recent multi-centre,
Structural Epilepsy

Fig. 5.13. MRI of the brain of a 2-year 5-month-old, male, English springer spaniel with traumatic brain injury following a horse kick. The dog had a seizure shortly after the head trauma. The MGCS was 10 on presentation a few hours after the injury. Dorsal (a), sagittal (b), transverse (c) T2W and transverse T1W (d) and T2* GRE (e) images show a comminuted fracture of the right frontal sinus and of the frontal bone extending into the parietal bone. The right frontal sinus is filled with heterogeneous material, mostly hypointense on T2* GRE (e) suggesting haemorrhage. Some of the frontal bone fragments are depressed. There are multiple intraparenchymal haemorrhages and diffuse oedema within the right forebrain. There is extensive muscle oedema surrounding the right side of the calvarium.

controlled trial, treatment focused on maintaining monitored ICP at 20 mm Hg or less was not shown to be superior to treatment based on imaging and clinical examination (Chesnut et al., 2012).
Treatment
The majority of therapeutic recommendations for animals with TBI are based on human head trauma studies and experimental investigations due to a lack of retrospective or prospective veterinary clinical studies (Dewey, 2000). Medical treatment of TBI starts soon after the initial assessment and primarily involves oxygen therapy, management of ventilation, fluid therapy and support of MABP (see Table 5.13). The primary goal of fluid therapy (including isotonic crystalloids, hypertonic crystalloids, artificial colloids and blood products) in patients with TBI is rapid restoration of normotension to ensure adequate CPP. Initial extracranial stabilization is closely followed by therapies directed toward intracranial stabilization including facilitating cerebral venous drainage, administration of hyperosmotic agents (in animals with MGCS <8) and AEMs (in seizuring animals). Cerebral venous drainage can be increased (without deleterious changes in cerebral oxygenation) by positioning the patient on a stiff board tilted at a 15–30° angle from the horizontal. The cervical spine must be manipulated carefully until fracture or luxation is ruled out and jugular vein compression should be avoided. These simple measures can help to minimize increases in ICP. Hyperosmotic agents such as mannitol and hypertonic saline can be used to decrease ICP and improve CPP and cerebral blood flow in animals with severe (MGCS <8) or deteriorating neurological dysfunction following TBI. Mannitol can be administered to normovolaemic patients at a dose of 0.5 to 1.5 g/kg as a slow intravenous bolus over 15–20 min (Sande, 2010). Boluses can be repeated (usually up to three times in 24 h) depending on patient response and initial dosage. Electrolytes should be monitored. Mannitol administration should be followed with
Structural Epilepsy
appropriate crystalloid therapy to prevent dehydration and hypotension, correct any electrolyte abnormality and maintain osmolality within normal range (290–310 mOsm/kg). Hypertonic saline can be used as an alternative to mannitol (particularly in hypovolaemic animals) or in animals that have already received multiple mannitol doses. Hypertonic saline can be administered intravenously over 2–5 min at a dose of 4 ml/kg of 7.5% sodium chloride or 5.3 ml/kg of 3% sodium chloride. Hypertonic saline is contraindicated in chronically hyponatraemic (see hyponatraemia, Chapter 4) as well as in hypernatraemic patients (Sande, 2010). The use of corticosteroids, including methylprednisolone, is currently not recommended in dogs and cats with TBI based on the lack of evidence for any beneficial effect and on results of clinical trials in humans showing increased mortality in TBI patients administered corticosteroids (Sande, 2010). Decompressive craniectomy is indicated in animals that do not improve or deteriorate neurologically despite aggressive medical therapy, extra-axial haematoma, open or depressed calvarial fractures, potentially contaminated bone fragments or foreign material within the brain parenchyma, ventricular obliteration and mass effect identified on advanced brain imaging. Anti-epileptic treatment is presented in the section on post-traumatic seizures and epilepsy. For further details on all the aspects of TBI treatment the reader is directed to more comprehensive descriptions (Dewey, 2000; Sande, 2010; Freeman and Platt, 2012).
Prognosis
Prognosis depends on severity of neurological dysfunction, concurrent injuries or complications and response to treatment. The MGCS predicts the probability of survival in the first 48 h after head trauma in an almost linear fashion with a 50% probability of survival in dogs with a score of 8 (Platt et al., 2001). In addition, there is a linear trend between the MGCS and survival at 1 and 6 months (Platt et al., 2007). The degree of hyperglycaemia is associated with severity of head trauma, however it does not seem to be associated with outcome in dogs and cats with head trauma. Because hyperglycaemia can potentiate neurologic injury, iatrogenic hyperglycaemia should be avoided in patients with head trauma (Syring et al., 2001). Severity of MRI changes (graded I–IV) in dogs with TBI significantly associates with outcome at 1 and 6 months (Platt et al., 2007).
Anomalous and Developmental
Malformations of the CNS can result from abnormalities of morphogenesis such as defects in neural induction, proliferation, differentiation and migration. These events are genetically regulated and interdependent. Underlying aetiologies involve genetic and environmental factors including in utero infection (often viral), toxicity, hypoxia-ischaemia or trauma. The gestational age of the insult affects the type and severity of the resultant malformation. Common malformations of the forebrain and associated clinical signs are listed in Table 5.14. Hydrocephalus is the most common developmental disorder that causes seizures and is discussed further in the text. Malformations of other areas of the CNS as well as CNS embryogenesis and development are beyond the scope of this text and the suggested reference can be consulted for additional information (DeLahunta and Glass, 2009).
Clinical signs
Clinical signs of a CNS malformation in dogs and cats are usually present from a young age although some malformations may not cause neurologic dysfunction until adulthood, if at all.
Diagnostic investigations
The majority of CNS malformations can be visualized on MRI. The MRI features of CNS malformations in dogs and cats have been described (Hecht and Adams, 2010a; MacKillop, 2011; Schmidt et al., 2012). Some malformations such as cerebral neuronal migration disorders
(e.g. heterotopias or dysplasias) may be diagnosed only upon histological examination of affected brain tissue (Plates 11 and 12). The majority of these cerebral neuronal migration disorders can result in seizures as the only neurological
Table 5.14. Malformations of the forebrain (DeLahunta and Glass, 2009b; MacKillop, 2011; Davies et al., 2012; Schmidt et al., 2012).
Onset of clinical Reported clinical signs in dogs Breed in which it has been Malformation Description signs and cats reported
Hydrocephalus (congenital) (see text for more information)
Hydranencephaly
Porencephaly
Meningoencephalocele
Excessive accumulation of CSF within cranial cavity (in the ventricular system and/or subarachnoid space)
Failure of development and destruction (secondary atrophy) primarily of the neocortex and the ventricular zone of the telencephalon resulting in a CSF-filled cavity without any residual brain parenchyma other than a thin layer of ependymal lining the expanded lateral ventricles. Usually the olfactory lobe, hippocampus and the basal nuclei are spared. The cranial cavity is normal in size and shape
Incomplete development and destruction (secondary atrophy) of primarily the neocortex and the ventricular zone of the telencephalon resulting in single or multiple CSF-filled cavity/ies within the brain that communicate/s with the ventricular system or subarachnoid space
Protrusion of brain tissue and meninges covered by skin through a defect in the skull (cranium bifidum or cranioschisis)
Usually within few to several weeks after birth
Within several weeks after birth
Within several weeks after birth or later in life
Commonly detectable from birth
Enlarged, dome-shaped cranium, mechanical bilateral ventrolateral strabismus, abnormal mentation, behavioural abnormalities, visual and auditory deficits, seizures, ataxia, circling and head pressing
Abnormal mentation, behavioural abnormalities, circling, visual deficits, seizures, difficulty or inability to suckle or to prehend food and drink water
Seizures only or with concurrent behavioural abnormalities; sometimes there are no clinical signs
Abnormal mentation, behavioural abnormalities and seizures Toy and brachycephalic breeds of dog
Miniature poodle, Labrador, mastiff cross, cavalier king charles spaniel, Lhasa apso, kittens of any breed with in utero feline parvovirus infection (feline panleukopaenia virus)
Beagle, collie cross, cavalier king charles spaniel, Australian shepherd, golden retriever, Jack Russell terrier, mixed breed dog, domestic shorthair and oriental shorthair cat
Burmese kittens (autosomal recessive inheritance; associated with other craniofacial malformations), any feline breed following exposure of pregnant cats to a variety of teratogenic agents including griseofulvin. German shepherd dog (frontoethmoidal meningoencephalocele)
160 L. De Risio
Meningocele
Exencephaly Holoprosencephaly
Agenesis or dysgenesis of the corpus callosum
Lissencephaly
Polymicrogyria
Cerebral neuronal heterotopias or dysplasias
Protrusion of a fluid-filled sac of meninges without any brain parenchyma through a defect in the skull
Protrusion of brain tissue and meninges (not covered by skin) through a defect in the skull
Failure of the forebrain to separate into two discrete cerebral hemispheres. Has been classified as alobar (arrhinencephaly), semilobar and lobar depending on the severity of the malformation. In all three forms, the corpus callosum fails to develop. Can be associated with cyclopian malformation
Incomplete or failure of development of the corpus callosum. It usually is accompanied by other defects
Disorder of cortical neuronal migration in which the developing brain fails to form the normal surface convolutions (gyri and sulci), giving the cerebral surface a smooth appearance. This can result in agyria, a complete absence of gyri; or in pachygyria, an abnormally increased thickness of the cerebral cortex with reduction in the number of gyri and abnormal laminar pattern of organization, with only four discernable layers rather than the normal six
Disorder of cortical neuronal migration resulting in increased numbers of small, disorganized gyri with abnormal cortical architecture histologically; may be generalized or lobar in distribution but consistently affects the occipital lobes in dogs. May occur concurrently with hydrocephalus
Disorder of cortical neuronal migration resulting in disorganized grey matter in inappropriate locations
Commonly detectable from birth
Detectable from birth
Soon after or within a few weeks after birth
Within a few weeks after birth
Usually in the first year of life
Commonly within the first 6 months of life
About 7 weeks of age (Rusbridge and Wilkins, 2002)
Hypodipsic hypernatraemia and behavioural abnormalities (such as inability to house-train), abnormal mentation and seizures
Hypodipsic hypernatraemia, abnormal mentation and behaviour
Abnormal mentation, behavioural abnormalities, visual deficits, delayed postural reactions and seizures
Abnormal mentation, behavioural abnormalities, central blindness or visual deficits, ataxia and seizures
Facial dysmorphism, tetraparesis, hypermetria and intention tremor (Rusbridge and Wilkins, 2002)
Pomeranian Miniature schnauzers
Miniature schnauzers, kittens born from a queen exposed to griseofulvin
Lhasa apso dogs, wire-haired fox terriers, Irish setters, and samoyeds (in combination with cerebellar hypoplasia and dysplasia), and in Korat cats (with associated microencephaly)
Standard poodles
Lagotto Romagnolo (Rusbridge and Wilkins, 2002), epileptic dog (Buckmaster et al., 2002)
Structural Epilepsy
abnormality and have been shown to represent the underlying aetiology of a substantial number of focal epilepsies in humans. Heterotopic cell clusters have also been reported in one epileptic dog (Buckmaster et al., 2002).
Treatment
Treatment of a CNS malformation is symptomatic. Anti-epileptic treatment is performed as for other types of structural brain disorders (see introduction to this chapter and Chapters 12–24).
Prognosis
Prognosis is variable depending on type of malformation, severity of neurological dysfunction and response to treatment.
Hydrocephalus
Pathophysiology and classification
Hydrocephalus is characterized by excessive accumulation of cerebrospinal fluid (CSF) within the cranial cavity with subsequent dilation of the ventricular system or subarachnoid space (Plate 13).
Table 5.15. Classification of hydrocephalus.
CSF is produced by the choroid plexuses of the lateral, third and fourth ventricles, parenchymal capillaries and leptomeningeal capillaries in the subarachnoid space. CSF production is independent of hydrostatic pressure within the ventricles but is influenced by osmotic pressure of the blood, as is demonstrated by the fact that intravenous administration of hypertonic solutions reduces the rate of formation of CSF. CSF flows from the lateral ventricles through the interventricular foraminae to the third ventricle. It continues caudally through the mesencephalic aqueduct to the fourth ventricle and into the subarachnoid space through the lateral apertures of the fourth ventricle. The CSF is absorbed mainly by arachnoid villi in the venous sinuses and cerebral veins and to a lesser extent by venous and lymphatic drainage around spinal and cranial nerves (DeLahunta and Glass, 2009).
Hydrocephalus has been classified in different ways according to its anatomic relationship with the ventricular system and subarachnoid space (internal or external), to the underlying disease process (obstructive, non-obstructive or compensatory), development prior to or after birth (congenital or acquired) and according to pressure differences (normotensive, hypertensive) (Table 5.15) (Coates et al., 2006; Thomas, 2010).
| Type of classification | Type of hydrocephalus | Definition |
| Location of excessive | Internal | Excessive CSF accumulation within the ventricular |
| CSF accumulation | External | space Excessive CSF accumulation within the intracranial |
| Underlying disease process Occurrence prior or after birth Pressure differences | Obstructive (non-communicating) Non-obstructive (communicating) Compensatory (ex vacuo) Congenital Acquired Normotensive Hypertensive | subarachnoid space Obstruction of CSF flow within the ventricular system, subarachnoid space or between the ventricular system and the subarachnoid space, or decreased CSF absorption Increased production of CSF CSF occupies space in the cranial cavity that normally would be occupied by brain parenchyma, absent due to destruction or lack of development Present at birth Acquired after birth Normal CSF pressure Increased CSF pressure |
Structural Epilepsy
Congenital hydrocephalus is considered more common than acquired hydrocephalus in veterinary patients (Coates et al., 2006). The most commonly identified cause of congenital hydrocephalus has been mesencephalic aqueductal stenosis associated with fusion of the rostral colluliculi resulting in obstructive hydrocephalus affecting the lateral and third ventricles and, occasionally, the cavity of the olfactory bulb and infundibular recess. However, an obvious site of obstruction is often not apparent in clinical practice. In these cases, hydrocephalus may be due to obstruction at the level of the subarachnoid space, arachnoid villi or lateral apertures. Alternatively, intraventricular obstruction could occur during a critical stage of development and subsequently resolve, leaving only the ventricular enlargement (Thomas, 2010). Congenital hydrocephalus may be associated with several other anomalies of the CNS (DeLahunta and Glass, 2009). Congenital hydrocephalus is most common in toy and brachycephalic breed dogs. Maltese, Yorkshire terrier, English bulldog, Chihuahua, Lhasa apso, Pomeranian, toy poodle, Cairn terrier, Boston terrier, pug and Pekingese have been reported at increased risk for congenital hydrocephalus (Selby et al., 1979).
Clinical signs
Clinical signs of congenital hydrocephalus in dogs and cats are usually recognized at a very young age. Animals with congenital hydrocephalus are often smaller than their litter-mates. If the increase in intracranial volume occurs before the sutures of the calvaria have closed, the cranial cavity will be enlarged, dome-shaped (Fig. 5.14) with open sutures and persistent fontanelles. Bilateral ventrolateral strabismus may occur secondary to mechanical pressure on the eyes from orbital malformation or anatomic alterations of the midbrain and oculomotor nuclei (Fig. 5.15) (Coates et al., 2006). Neurologic deficits include abnormal behaviour, cognitive dysfunction such as inability to become house trained, altered mental status, ataxia, circling, head pressing, visual deficits, seizures, and vestibular dysfunction (Thomas, 2010). In a recent study, seizures were reported in 27% (8/30) of dogs and 67% (4/6) of cats with congenital internal hydrocephalus (Biel et al., 2013). Seizures occurred concurrently with other forebrain signs in all but one animal (Biel et al., 2013). Papilloedema may be seen on fundic examination in a small percentage of cases.

Hydrocephalus can lead to abnormal hypothalamic function. Hypodipsic hypernatraemia, possibly as a result of pressure atrophy of hypothalamic osmoreceptors, has been reported (Dow et al., 1987; DiBartola et al., 1994). The course of disease is variable and difficult to predict. Neurologic deficits can progress over time, remain static, or even improve after 1 to 2 years of age (Thomas, 2010).
Acquired hydrocephalus can develop at any age secondary to diseases such as meningoencephalitis or neoplasia. Neurologic deficits are similar to those described for congenital hydrocephalus and may also reflect the underlying cause of the hydrocephalus.
Diagnostic investigations
Diagnosis is based on the clinical signs and brain imaging to assess ventricular size and identify any possible underlying causes of hydrocephalus (Thomas, 2010). Symmetric or asymmetric enlargement of the lateral ventricles can occur in clinically normal juvenile and adult dogs, therefore the diagnosis of hydrocephalus must be based on the combination of clinical and imaging findings, not just ventricular size. Ventricular size, symmetry and volume have been reported in a few canine breeds and in kittens using MRI and ultrasound (US) (Spaulding and Sharp, 1990; Haan et al., 1994; Kii et al., 1997; Vite et al., 1997; Esteve-Ratsch et al., 2001; Jaderlund et al., 2003).

Radiographic signs suggestive of hydrocephalus include doming of the calvarium with thinning of cortical bone, decreased prominence of normal calvarial convolutions, and persistent suture lines and fontanelles. Ultrasonography can be performed in animals with persistent bregmatic fontanelles and allow detection of lateral ventricle enlargement. These can be seen as paired anechoic regions (Fig. 5.16) or as a single, large anechoic structure when the septum pellucidum (that normally separates the lateral ventricles) is absent. Optimal resolution is provided by a high-frequency probe (7–12 MHz). The main benefit of ultrasonography is that it can generally be performed without the need for sedation or general anaesthesia. CT and MRI enable accurate assessment of the entire ventricular system, subarachnoid space, extent of cortical atrophy and can help to identify the underlying causes of hydrocephalus (e.g. stenosis of the mesencephalic aqueduct, a mass obstructing CSF flow or meningoencephalitis). Obstructive hydrocephalus is commonly associated with ventricular dilatation proximal to the obstruction site, and with preservation of normal ventricular size distal to the block (Fig. 5.17). MRI is more sensitive than CT in demonstrating parenchymal changes,

Fig. 5.16. Transverse ultrasound image of the brain via the bregmatic fontanelle of a 5-month-old male Chihuahua with progressive forebrain signs. Note the severely enlarged lateral ventricles (LV).
Structural Epilepsy
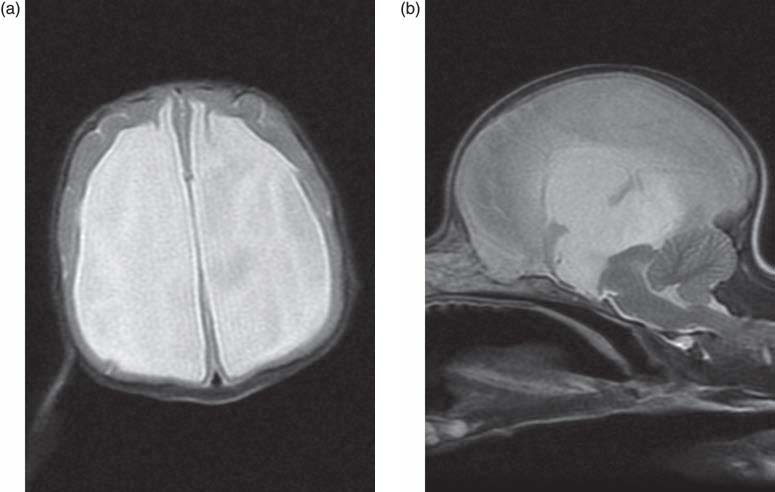

especially in the caudal fossa. Periventricular white matter interstitial oedema resulting from trans-ependymal flow of CSF due to increased intraventricular pressure may be identified in some animals with hydrocephalus and appear as a rim of hyperintensity surrounding the ventricular system on T2W and fluid attenuated inversion recovery (FLAIR) MR images.
Analysis of the CSF is helpful in cases of hydrocephalus associated with suspected meningoencephalitis. As with other disorders that can result in increased ICP, CSF collection should be performed following brain MRI or CT to minimize the risks associated with the procedure.
Treatment
The treatment of hydrocephalus depends on the cause of the disorder and the status of the animal. Mildly affected and clinically stable animals may not require any treatment (e.g. those with compensatory hydrocephalus). Severely affected animals (e.g. those with acute obstructive hydrocephalus) with increased ICP would need emergency medical treatment to decrease cerebral oedema (see treatment of traumatic brain injury) and CSF production (see medical treatment of hydrocephalus). This may be followed by surgery. In animals with acquired hydrocephalus, the underlying cause needs to be treated medically and/or surgically whenever possible (e.g. meningitis, meningoencephalitis, neoplastic or granulomatous mass obstructing CSF flow). As most animals with congenital hydrocephalus have obstructive hydrocephalus, medical therapy is mainly palliative, as it does not relieve the obstruction. The main goal of medical treatment is to provide clinical improvement with the least possible amount of medication. Acetazolamide, a carbonic anhydrase inhibitor, can be used alone or in combination with furosemide, to decrease CSF production. Acetazolamide is started at 10 mg/kg orally every 8 h. Furosemide can be added at 1 mg/kg orally once daily. The dose is tapered based on clinical effect. Acetazolamide should be used short term. Electrolyte, acid-base status and hydration shouldbemonitored (Thomas, 2010). Alternatively, omeprazole (0.7 mg/kg/day, or 10 mg/day for dogs <20 kg or 20 mg/day for dogs >20 kg), H+/K+ pump inhibitor can be used alone or in combination with other medications to reduce CSF production. Glucocorticoids have also been used to treat hydrocephalus in veterinary patients at 0.25 to 0.5 mg/kg twice daily until signs improve, then the dosage is reduced at weekly intervals down to 0.1 mg/kg every other day. Anti-epileptic treatment is performed as for other types of structural brain disorders (see introduction to this chapter and Chapters 12–24).
Surgical treatment is reserved for animals with progressive neurological dysfunction refractory to medical therapy, however it should be instituted before neurological deficits become permanent. The goal of surgical treatment is to halt disease progression and improve the neurologic status of the animal (Coates et al., 2006). Surgical treatment is most commonly used in animals with congenital obstructive hydrocephalus, and sometimes in animals with acquired hydrocephalus that require rapid CSF drainage to decrease ICP. Surgery is not indicated in animals with systemic, abdominal or skin infection at the site of the cranial or abdominal incision; or in whom a successful surgery would not change the outcome, such as an animal with multiple, severe brain malformations (Thomas, 2010). Surgical treatment consists of placement of a ventriculoperitoneal shunt, a CSF drainage device that diverts CSF from the lateral ventricle to the peritoneal cavity. A shunt system consists of a ventricular catheter, a one-way pressure valve and a distal catheter that carries CSF from the valve to the peritoneal cavity. Low or ultralow pressure valves should be used in dogs and cats. The shunt tubing may need to be replaced as the animal grows. The surgical technique for ventriculoperitoneal shunt placement has been described (Coates et al., 2006; Thomas, 2010; de Stefani et al., 2011). The major disadvantages of surgery are the expense and the post-operative complications. Complications include occlusion of the shunt catheter by fibrous tissue, clot debris, choroid epithelium or ependyma infection; kinking, breaking or disconnection of catheter components; over- or under-drainage; wound dehiscence,
Structural Epilepsy
skin breakdown over shunt hardware, CSF fistula formation, pain and seizures (Coates et al., 2006; Thomas, 2010; de Stefani et al., 2011; Shihab et al., 2011; Biel et al., 2013). Complications may be readily treated medically or surgically. Complication rate following ventriculoperitoneal shunt implantation ranges from 29 to 50% (de Stefani et al., 2011; Shihab et al., 2011; Biel et al., 2013). Ventriculoperitoneal shunt implantation can result in short- to long-term neurological improvement, however blindness rarely resolves and seizure activity may persist (Coates et al., 2006; de Stefani et al., 2011; Shihab et al., 2011; Biel et al., 2013).
Prognosis
Prognosis depends on the cause of hydrocephalus, severity of neurological dysfunction and response to treatment. Even with successful surgical treatment, a realistic expectation is improvement rather than complete resolution of neurologic dysfunction. In children with hydrocephalus, epilepsy has an incidence of 6–30% and is associated with a poor outcome (Vinchon et al., 2012).
Neoplastic
Neoplasia of the nervous system can be classified as primary and secondary. Primary nervous system tumours originate from neuroectodermal, ectodermal and/or mesodermal cells normally present in or associated with brain, spinal cord or peripheral nerves. Secondary tumours affecting the nervous system may originate from haematogenous metastasis of a primary tumour in another organ or from structures surrounding the neuroparenchyma, such as the nose, ear, calvaria, pituitary gland or vertebrae and may affect the neural tissue by infiltration or compression. Dissemination or metastasis of CNS tumours is rare, but may occur via the CSF pathways, especially if tumours are located close to the subarachnoid space or ventricular cavities (e.g. choroid plexus tumours or ependymoma), or via a haematogenous route, such as the dural sinus, with subsequent development of distant metastasis, usually in the lung (Vite, 2005). This section will focus on intracranial neoplasia.
Intracranial neoplasia
Intracranial tumours exert their pathologic effects both by compressing and/or infiltrating the neuroparenchyma and by secondary effects such as peritumoural oedema, inflammation, obstructive hydrocephalus and haemorrhage.
Canine and feline intracranial neoplasias are listed in Box 5.5. The most common intracranial tumour types in dogs and cats are discussed in the text below.
Incidence
An accurate estimate of the incidence of intracranial neoplasia in animals is unknown. One study reported a rate of 14.5 and 3.5 per 100,000 dogs and cats, respectively (Vandevelde, 1984). In other studies, the incidence of
Box 5.5. Canine and feline intracranial neoplasias.
Primary:
• Primitive neuroectodermal tumours (neuroblastomas, medulloblastoma, gangliocytomas);
histiocytosis).
Secondary:
prostatic, pancreatic, pulmonary);
• Nasal tumours (e.g. adenocarcinoma, squamous cell carcinoma, chondrosarcoma, neuroesthesioblastoma);
• Histiocytic sarcoma;
• Calvarial osteosarcoma and multilobulated tumour of bone (multilobulated osteochondrosarcoma);
intracranial neoplasia was 2.8% and 2.2% in dogs and cats, respectively (Zaki and Hurvitz, 1976; Zaki, 1977). The most commonly reported primary brain tumour in dogs and cats is meningioma (Troxel et al., 2003; Snyder et al., 2006). The second most common primary brain tumours in dogs and cats are astrocytoma and oligodendroglioma (Troxel et al., 2003; Snyder et al., 2006). The most common secondary tumour is haemangiosarcoma in dogs and lymphoma in cats (Troxel et al., 2003; Tomek et al., 2006; Snyder et al., 2008). Pituitary tumours are the second most common secondary tumour in dogs and cats (Troxel et al., 2003; Snyder et al., 2008). The golden retriever and boxer have been reported at increased risk to develop intracranial tumours (primary and secondary) (Snyder et al., 2006; Sturges et al., 2008). Meningiomas have been most commonly reported in dolicocephalic dog breeds, especially German shepherds, golden retrievers and Labrador retrievers as well as in the boxer (Snyder et al., 2006; Sturges et al., 2008). Glial tumours (astrocytomas and oligodendrogliomas) have been reported in the boxer, Boston terrier and other brachycephalic breeds with higher frequency than in other canine breeds (Snyder et al., 2006). In addition to the golden retriever and boxer, Labrador retrievers and German shepherd dogs are most commonly affected by secondary intracranial tumours (Snyder et al., 2008). The reported mean age of dogs and cats with primary brain tumours is
9.5 and 11.3 years, respectively (Bagley et al., 1999; Troxel et al., 2003; Snyder et al., 2006). Dogs and cats with meningioma are significantly older at diagnosis than those with other primary brain tumours (Troxel et al., 2003; Snyder et al., 2006). The mean age of dogs with secondary brain tumours is 9.6 years (Snyder et al., 2008). The mean age of cats with secondary brain tumours is the same as for primary brain tumours (Troxel et al., 2003). No gender predisposition for different intracranial tumour types has been consistently reported.
Clinical signs
Clinical signs commonly reflect the location and the secondary effects of the tumour (e.g. cerebral oedema, obstructive hydrocephalus, haemorrhage, infarction, bacterial infection and inflammation in case of communication with the nasal cavity and brain herniation). Onset of signs is generally chronic and progressive, however acute onset or deterioration can occur with haemorrhage, obstructive hydrocephalus, or sudden exhaustion of cerebral compensatory mechanisms resulting in severely increased ICP (see section on TBI for clinical signs and management of increased ICP). Seizures, behavioural changes and altered mentation have been reported as the most common presenting signs in dogs and cats with primary and secondary supratentorial intracranial neoplasia (Troxel et al., 2003; Snyder et al., 2006; Schwartz et al., 2011; Rossmeisl et al., 2013). Seizure can be the only clinical abnormality in animals with tumours localized to the olfactory bulb or frontal lobes (Foster et al., 1988; Smith et al., 1989). Central vestibular signs are common in dogs with caudotentorial intracranial tumours (Rossmeisl et al., 2013).
Tumour-related seizures and epilepsy
Prevalence of seizures in animals with intracranial neoplasia varies among studies ranging from 45% (Bagley et al., 1999) to 73% (Rossmeisl et al., 2013) in dogs and nearly 23% in cats (Troxel et al., 2003; Tomek et al., 2006). Seizures can occur as single events, cluster seizures or less commonly as status epilepticus (Tomek et al., 2006; Schwartz et al., 2011). Seizures can be the only clinical abnormality when the neoplasia is focally affecting the so-called ‘clinically silent regions’, such as olfactory bulb, frontal and pyriform lobes. Risk factors for development of seizures associated with intracranial neoplasia are magnetic resonance imaging findings consistent with the presence of neoplastic tissue in frontal lobe, subfalcine and/ or subtentorial herniation and marked gadolinium enhancement of the tumour (Schwartz et al., 2011).
In people with brain tumours, seizures are the presenting symptom in 20–50% of patients, and occur during the course of the oncological disease in a further 20–45% of patients (Maschio, 2012; You et al., 2012). Similarly to what has been reported in dogs, the overall incidence of epilepsy in people with brain tumours (regardless of histological type and
Structural Epilepsy
anatomical site of the lesion) varies from 35 to 70% (Maschio, 2012). Tumours in the frontal and temporal lobes, as well as in the limbic system, are more likely to cause seizures than tumours in other locations (You et al., 2012).
The pathogenesis of tumour-related epilepsy remains poorly understood. Both tumoural and peri-tumoural factors could contribute to epileptogenesis by causing changes in neurotransmitters and their receptors, extracellular ions, perfusion, metabolism, and immunological and inflammatory responses (You et al., 2012).
Tumoural factors include:
inhibitory and anti-epileptogenic effects) in gliomas; and
• Immunological or inflammatory changes.
Anti-epileptic treatment should be instituted in all animals with seizures caused by intracranial neoplasia. The choice of the AEM is affected by many variables including severity and frequency of seizures, presence and degree of neurological deficits (e.g. decreased mental status and ataxia), hepatic and renal function, interaction with other medications (e.g. chemotherapeutic agents), owner life style and costs (see Chapter 12). Phenobarbital is commonly used in animals with normal hepatic function and with normal to mildly obtunded mental status. Loading may be necessary when rapid achievement of target levels is required, however this could affect monitoring of neurological status by causing or worsening sedation and ataxia. In a prospective study on palliative treatment of primary brain tumours, 70% (21/30) of dogs on phenobarbital developed or had exacerbations of existing sedation or ataxia after starting the treatment (Rossmeisl et al., 2013). Alternatively, levetiracetam may be used to control seizures at least initially. Potassium bromide or phenobarbital at maintenance dosage may be started at the same time as levetiracetam to provide longer-term seizure control while avoiding the undesirable effects of loading. Alternatively, a new generation AEM such as zonisamide or levetiracetam may be preferable as initial monotherapy in animals with moderately to severely obtunded mental status prior to antiepileptic treatment or as a substitute to phenobarbital or potassium bromide in animals with persistent sedation due to these old-generation AEMs.
New-generation AEMs may also be preferable in animals needing lifelong corticosteroid administration as adverse effects of phenobarbital and potassium bromide (e.g. polyuria, polydipsia and polyphagia) are likely to be compounded by concurrent steroid use. In one study, one-third of dogs with supratentorial tumours developed epilepsy refractory to phenobarbital treatment and this was the most common manifestation of tumour progression that prompted euthanasia (Rossmeisl et al., 2013).
In people, brain tumour-related epilepsy is often drug-resistant, has a strong impact on the quality of life, and is considered the most important risk factor for long-term disability (Maschio, 2012). Therefore selecting the most effective and well-tolerated AEM is very important. However, as in veterinary medicine the choice of AEM in brain-tumour epileptic patients is based on the neurologist’s preference due to the lack of randomized controlled clinical evidence (Kerrigan and Grant, 2011). New-generation AEMs such as levetiracetam, zonisamide and lacosamide are preferred because they have fewer pharmacokinetic interactions and cause fewer adverse effects (Maschio, 2012). In particular, levetiracetam has no significant pharmacokinetic interactions with chemotherapy agents and has been shown to be very effective and well tolerated for both monotherapy and adjunct therapy in people with tumour-related epilepsy (Kerrigan and Grant, 2011). AEM-resistance in patients with brain tumourrelated epilepsy may be caused by changes in the tumoural and peri-tumoural biochemical milieu, pharmacokinetic interactions between AEMs and chemotherapeutics, over-expression of multidrug resistance-related proteins in tumours, which restrict the penetration of lipophilic substances into the brain, as well as tumour progression. Prophylactic anti-epileptic therapy in people with brain tumours is not routinely recommended due to lack of efficacy in preventing seizure onset and the potential serious adverse effects (Maschio, 2012). For management of tumourrelated status epilepticus, see Chapter 24.
Diagnostic investigations
The diagnostic investigation of animals with suspected intracranial neoplasia includes haematology, serum biochemistry profile, urinalysis, thoracic radiography and abdominal ultrasonography to investigate for concurrent extracranial disease including primary and metastatic neoplasia, brain imaging, CSF analysis, fine needle aspirate, cytology, biopsy and histology. In addition echocardiography is indicated in case of suspected haemangiosarcoma or cardiac dysfunction. Thoracic radiographic abnormalities such as metastatic lesions or concurrent disease (e.g. pneumonia, mega-oesophagus, heart failure) have been detected in approximately 20% of dogs with primary intracranial neoplasia and in 54% of dogs with secondary intracranial neoplasia (Snyder et al., 2006, 2008). Neoplasia unrelated to the primary intracranial tumour, mostly involving the thoracic or abdominal cavity, has been detected at necropsy in 23% (38/170) of dogs in one study (Snyder et al., 2006). Fine needle aspirate or true cut biopsy of any accessible extraneural masses may help to reach a definitive diagnosis of primary or metastatic neoplasia related or unrelated to the primary intracranial tumour. MRI or CT of the brain can help to support the ante-mortem diagnosis of intracranial neoplasia and particularly MRI can often provide a relatively accurate presumptive diagnosis. Meningioma has been correctly diagnosed based on MRI findings in 100% of dogs and 95% of cats with histologically confirmed intracranial tumours (Polizopoulou et al., 2004; Troxel et al., 2004). The MR imaging features of primary and common secondary canine and feline intracranial neoplasia have been described and are summarized in Table 5.16 (Troxel et al., 2004; Westworth et al., 2008; Hecht and Adams, 2010b; Palus et al., 2011; Ródenas et al., 2011; Wisner et al., 2011; Young et al., 2011; Martin-Vaquero et al., 2012). Some non-neoplastic intracranial lesions can present with imaging features similar to intracranial neoplasias. Sensitivity and specificity of high-field MRI for classifying brain diseases as neoplastic are 87.4 and 91.7% without provision of clinical data and 90.6 and 81.9% with provision of clinical data, respectively (Wolff et al., 2012). The estimated sensitivity and specificity for detecting specific neoplastic brain diseases varies greatly depending on tumour type. Sensitivity ranges from 0% (without or with provision of clinical data) for lymphoma to 84.4 and 91.1% without or with provision of clinical data, respectively, for gliomas. Specificity without or with provision of clinical data is approximately 98% for lymphoma, pituitary tumours and nasal adenocarcinomas, and 94% for meningioma, glioma, choroid plexus tumours (Wolff et al., 2012). In addition, MRI allows identification of the secondary pathophysiological effects caused
Table 5.16. The MR imaging features of primary and common secondary intracranial neoplasia in dogs and cats (by convention, the terms hypo-, hyper- and iso-intense describe signal intensity relative to that of normal grey matter in the same image).
Intensity on Intensity on Contrast enhancement Peritumoural Neoplasia Axial origin Anatomic features T1W images T2W images (T1WC images) oedema
| Meningioma | Extra-axial | Round/ovoid or plaque-shaped; |
| occasionally cystic; smoothly | ||
| marginated, broad-based | ||
| external margin conforming to | ||
| the meningeal plane; expansile | ||
| rather than infiltrative growth | ||
| pattern; often associated with | ||
| thickening of adjacent | ||
| meninges (dural tail sign) | ||
| Astrocytoma | Intra-axial | Round to ovoid with distinct |
| margins; or amorphous mass | ||
| to diffuse infiltrate with poorly | ||
| defined margins. Can contact | ||
| the meninges or the ventricles. | ||
| Intratumoural haemorrhage and | ||
| cystic areas may occur | ||
| Oligodendro- | Intra-axial | Round to ovoid or irregularly |
| glioma | shaped with distinct to poorly | |
| defined margins; can contact | ||
| the meninges or the ventricles; | ||
| intratumoural haemorrhage and | ||
| cystic areas can occur | ||
| Gliomatosis cerebri | Intra-axial | Diffuse, widespread infiltration of |
| the CNS by neoplastic glial | ||
| cells with relative preservation | ||
| of the neural tissue architecture |
Usually uniformly isointense and occasionally hypo- or hyperintense
Isointense or mildly to moderately hypointense
Moderately hypointense
Isointense or mildly hypointense Common
| Usually hyperintense and less commonly isointense | Usually marked and uniform or heterogeneous |
| Moderately hyperintense | Absent to minimal in low grade astrocytomas and moderate or marked, nonuniform, or peripheral in high grade astrocytomas |
| Hyperintense | Highly variable ranging from none to intense and peripheral or nonuniform |
| Hyperintense | Absent |
Variable, usually minimal to moderate. Marked in grade IV astrocytoma (glioblastoma multiforme)
Minimal to moderate
Absent to minimal
Continued
Structural Epilepsy
Table 5.16. Continued.
Intensity on Intensity on Contrast enhancement Peritumoural Neoplasia Axial origin Anatomic features T1W images T2W images (T1WC images) oedema
Ependymoma
Choroid plexus tumours
Choroid plexus papillomas (CPP) and choroid plexus carcinomas (CPC)
Primitive neuroectodermal tumours (PNETs)
Metastatic haemangiosarcoma
Predominantly intraventricular
Intraventricular
Intra-axial Intra-axial
Well-circumscribed, smooth or lobulated mass associated with the ventricular system; may expand to fill the ventricular cavity in which they arise, causing distortion of the ventricle and obstructive hydrocephalus. May contain cysts and/or haemorrhage
Smooth, round or lobulated mass, it may conform to the shape of the ventricle in which they grow; may contain cyst(s), areas of mineralization, haemorrhage, or necrosis. CPP may have a grossly papilliform shape. Concurrent ventriculomegaly and periventricular oedema are common. Intraventricular or subarachnoid metastases have been reported in 35% of CPC but not in CPP
Ill-defined mass
Ill-defined mass lesion(s); commonly multiple mass lesions, but single lesions are possible. Frequently distributed within the grey–white matter interface. Intratumoural haemorrhage is common and best detected on T2-W* sequences as signal void (hypointensity)
Slightly hypointense to slightly hyperintense
Hypo-, iso- or hyperintense
Hypointense to isointense
Mixed
Moderately to markedly hyperintense
Usually hyperintense, sometimes isointense
Hyperintense
Mixed
Frequently marked; may be heterogeneous
Intense and homogeneous
Moderate to strong and heterogeneous
Variable and often peripheral
Absent or minimal unless the tumour invades the periventricular brain parenchyma or hydrocephalus causes periventricular interstitial oedema
Common (44% of CPP and 69% of CPC)
Present
May be marked
172 L. De Risio
| Lymphoma | Intra-axial or | Ill- or well-defined, mass-like or | Isointense to | Hyperintense or | Consistently present, | Minimal to moderate |
| extra-axial | diffuse, single or multicentric. A ‘dural tail sign’ has been reported in cats with | hypointense | isointense | but with variable degree and distribution | peritumoural oedema | |
| Histiocytic tumours | Intra-axial or extra-axial | extra-axial lymphoma Ill- or well-defined, mass-like or diffuse, single or multicentric. | Iso- to hypointense | Iso- to hyperintense | Common | Regional or diffuse |
| May occur as diffuse meningeal infiltration. A ‘dural tail sign’ has been reported in dogs with | ||||||
| extra-axial histiocytic tumours |
| Pituitary | Extra-axial | Well-circumscribed and expansile | Isointense | Mildly | Marked and uniform | Common |
| tumours | mass; may be cystic and/or | hyperintense | (may be nonuniform | |||
| haemorrhagic. Pituitary | in pituitary | |||||
| carcinomas show more invasive | carcinomas) | |||||
| growth than adenomas, and | ||||||
| may invade adjacent | ||||||
| basisphenoid bone and | ||||||
| pharynx. | ||||||
| Dynamic studies or specific thin- | ||||||
| slice sections might be | ||||||
| necessary to diagnose | ||||||
| microadenomas | ||||||
| (<10 mm in height) |
Structural Epilepsy
by the tumour (e.g. cerebral oedema, haemorrhage, infarction, obstructive hydrocephalus and brain herniation).
Definitive ante-mortem diagnosis of intracranial neoplasia requires histological examination following stereotactic or surgical brain biopsy. However, cerebrospinal fluid cytology may sometimes allow a diagnosis of lymphoma (Plate 14) (Troxel et al., 2003; Palus et al., 2011).
Unless neoplastic cells are detected, which is uncommon, CSF analysis provides limited information in the diagnosis of primary intracranial neoplasia. As intracranial tumours may cause an increased ICP, CSF collection should be performed only if considered safe following brain MRI or CT. In animals with primary intracranial neoplasia, CSF analysis may be normal, reveal increased protein concentration alone or in association with pleocytosis. In one study on canine primary intracranial neoplasia, CSF pleocytosis occurred in 58% of dogs and was most commonly a mixed cell population (Snyder et al., 2006). Neutrophilic pleocytosis has been reported in 19% to 25% of dogs with intracranial meningiomas (Bailey and Higgins, 1986; Dickinson et al., 2006).
Treatment and survival times
Treatment of intracranial neoplasia can be divided into palliative and definitive.
The aim of palliative treatment is to alleviate clinical signs by minimizing the secondary effects of the intracranial neoplasia (mainly vasogenic oedema). Prednisone
(0.5 to 1.0 mg/kg/day orally) may be effective at reducing endothelial permeability, vasogenic oedema and CSF production. If improvement is observed, the dosage may be gradually reduced to the lowest effective dosage to control neurologic signs. In animals with suspected or confirmed increased ICP, administration of intravenous corticosteroids, mannitol or hypertonic saline (see section on increased ICP and traumatic brain injury) can be also effective in rapidly reducing vasogenic oedema, decreasing CSF production and stabilizing the endothelial membrane. Anti-epileptic treatment is presented in the section on tumourrelated seizures. In animals with hydrocephalus of the lateral ventricles related to obstruction of and/or overproduction of CSF (e.g. with a choroid plexus tumour), ventriculoperitoneal shunting can help control intracranial pressure and improve clinical signs (see Hydrocephalus section) (de Stefani et al., 2011).
Median survival for dogs receiving palliative pharmacological treatment for brain tumours varies among studies, ranging from
0.5 to 2.3 months (Heidner et al., 1991; Rossmeisl et al., 2013). Survival time of dogs with infratentorial tumours is significantly shorter (median, 28 days; 95% CI, 19 to 68 days) than survival time of dogs with supratentorial tumours (median, 178 days; 95% CI, 119 to 270 days) (Rossmeisl et al., 2013). Survival times are influenced by tumour type and location, severity of neurological signs, response to treatment and the pet-owner’s willingness to continue therapy.
Definitive treatment consists of surgical removal/debulking, radiation therapy and chemotherapy, alone or in combination. Definitive treatment is commonly combined with palliative pharmacological treatment. Benefits of surgical treatment include removal of neoplastic tissue, decompression of the neuroparenchyma, decrease of ICP and provision of a sample for histologic diagnosis. Surgery is generally aimed at solitary noninvasive tumours located on or near the brain surface. A careful and complete surgical resection, an intensive anaesthetic monitoring and postoperative care are essential for a favourable outcome. Surgical approaches to the brain and technical details on intracranial surgery have been described and are beyond the scope of this text (Glass et al., 2000; Meij et al., 2002; Bagley, 2003a, b; Forterre et al., 2006, 2009; Barreau et al., 2010; Uriarte et al., 2011). Most data on outcome and survival following intracranial surgery involve meningiomas in both dogs and cats and are described in the section on treatment and survival times of meningiomas. Information on microsurgical trans-sphenoidal hypophysectomy is presented in the section on treatment of pituitary tumours.
Radiation therapy may be performed following incomplete surgical resection or as sole treatment when the intracranial tumour is not surgically accessible or surgery is not considered the best therapeutic
Structural Epilepsy
option. Radiation therapy protocols vary, but most involve administration of a total radiation dose of 46 to 48 Gy in 2.0 to 4.0 Gy fractions daily or every other day. The acute adverse effects of radiation include cerebral oedema and, possibly, a temporary increase in seizure activity. Brain oedema generally responds to corticosteroid therapy. Late effects of radiation can be seen months to years after therapy and are due to brain necrosis. Clinical signs of late radiation damage are often similar to the initially presenting neurologic signs, and differentiation between radiation-induced damage and tumour recurrence can be challenging. Late effects of radiation cannot be effectively treated (Brearley et al., 1999). The median survival time for dogs with brain tumours treated with radiation therapy alone ranges from 4.7 months to 23.3 months (Turrel et al., 1984; Heidner et al., 1991; Brearley et al., 1999; Spugnini et al., 2000; Bley et al., 2005) and has been reported to be over
43.6 months in dogs with pituitary tumours (Bley et al., 2005; Kent et al., 2007). This great difference in survival times is most likely due to differences in radiation therapy protocols, tumour types, size and location, and severity of neurological signs.
Stereotactic radiosurgery has been used to deliver a single fraction (10 to 15 Gy) of radiation to canine intracranial tumours (Lester et al., 2001). The median survival time of three dogs with intracranial tumours (two meningiomas and one oligodendroglioma) treated with stereotactic radiosurgery is 14.2 months (Lester et al., 2001).
There is limited information on the use of chemotherapy for primary brain tumours in dogs and cats. Chemotherapeutic agents include hydroxyurea for meningiomas, lomustine and carmustine for meningiomas and gliomas and temozolamide for gliomas. Information on hydroxyurea and lomustine are detailed in the section on treatment of meningiomas. Other chemotherapeutic agents currently used in humans may have a role in the treatment of primary brain tumours in dogs and cats. Secondary brain tumours such as lymphoma may be treated with multidrug protocols, which often include chemotherapeutic agents that cross the blood-brain barrier such as lomustine or cytosine arabinoside.
Definitive treatment of brain tumours may also contribute to seizure control. In people, the percentage of seizure-free patients after tumour surgery ranges from 65 to 82%. The most significant factors associated with seizure freedom are completeness of tumour resection and short pre-operative duration of brain tumour-related epilepsy. Chemotherapy contributes to decreasing seizure frequency in 50–65% of patients with 20–40% of seizure freedom. Radiation therapy has an efficacy on seizure control ranging between 40 and 100% depending on different radiation techniques (gammaknife, conventional radiation therapy, radiosurgery) (Rudà et al., 2010).
Prognosis
Prognosis for animals with intracranial neoplasia is dependent on numerous factors, including tumour type and grade, tumour location, severity of neurological deficits, type and response to treatment.
Meningioma
Meningioma arises from the meninges, specifically the cap cells covering the arachnoid granulations, particularly at the point where they project into the venous sinuses (Summers et al., 2005). Most canine meningiomas are adjacent to the calvaria and located in the olfactory/frontal region, the floor of the cranial cavity, the optic chiasm or the suprasellar and parasellar regions (Snyder et al., 2006; Sturges et al., 2008) (Plate 15). Common locations of feline meningiomas include the tela choroidea of the third ventricle and the supratentorial meninges (Troxel et al., 2003) (Plate 16).
Incidence
Meningioma is the most commonly reported primary brain tumour in dogs and cats (Troxel et al., 2003; Snyder et al., 2006). In large retrospective studies on histologically confirmed primary intracranial neoplasia, meningioma has been diagnosed in 45% (Snyder et al., 2006) of dogs and 85% (Troxel et al., 2003) of cats. In cats, meningioma may be found as an incidental finding (in 22.6% of cases), occur as multiple meningiomas (in 17.2% of cases), or concurrently with another intracranial neoplasm including lymphoma, pituitary tumour and sarcomas (in 14.0% of cases) (Troxel et al., 2003). In dogs, occurrence of multiple meningiomas or of meningioma concurrently to another intracranial tumour type is uncommon (McDonnell et al., 2007; Sturges et al., 2008; Espino et al., 2009). Commonly affected breeds include domestic short hair cats, boxers and dolicocephalic canine breeds, especially German shepherds, golden retrievers and Labrador retrievers (Snyder et al., 2006; Sturges et al., 2008). No consistent gender predisposition has been identified. The reported mean age at diagnosis of meningioma is 11.1 years in dogs and
12.2 years in cats, which is significantly older than the age at diagnosis of other primary brain tumours (Troxel et al., 2003; Snyder et al., 2006).
Clinical signs
Clinical signs commonly reflect the anatomic location and the secondary effects of the tumour. Onset of neurological signs is generally slowly progressive. The severity of clinical signs depends on the growth rate and location of the meningioma, peritumoural oedema, intracranial pressure and compensatory mechanisms within the brain. If normal compensatory mechanisms are exhausted
(i.e. decreased CSF and blood volume), ICP can rapidly increase and clinical signs can present with an acute onset or deterioration. Seizures and altered mentation have been reported as the most common presenting signs in dogs and cats with intracranial meningiomas (Troxel et al., 2003; Snyder et al., 2006; Schwartz et al., 2011). Interictal neurological examination may be normal. In cats with an intracranial meningioma, non-specific clinical signs (such as lethargy, inappetence or anorexia) not obviously referable to neurologic dysfunction have been reported in 21.5% of cats (Troxel et al., 2003).
Diagnostic investigations
Diagnostic investigations have been described in the general section on intracranial neoplasia and briefly include haematology, serum biochemistry, urinalysis, thoracic radiography, abdominal ultrasonography, brain MRI (Fig. 5.18a–e) or CT, cerebrospinal fluid analysis and histology following biopsy or surgical removal (Plates 17 and 18).
Treatment and survival times
Treatment of intracranial meningiomas can be divided into palliative (prednisolone and AEMs in seizuring animals) and definitive (surgery, radiation therapy, chemotherapy) (see general section on treatment of intracranial neoplasia).
Reported median survival time for animals receiving palliative pharmacological treatment for intracranial meningioma is 18 days in cats (Troxel et al., 2003) and 0.5 to 6 months in dogs (Turrel et al., 1984; Heidner et al., 1991; Rossmeisl et al., 2013).
Feline meningiomas are commonly well encapsulated and easily delineated from normal brain and complete surgical resection can be achieved. Post-operative mortality associated with craniotomy for meningioma removal in cats is reported to be approximately 19%, with anaemia being the most common complication (Gordon et al., 2004). Reported median survival times following surgical resection of single or multiple intracranial meningioma in cats range from 21.7 to 27 months (Gallagher et al., 1993; Troxel et al., 2003; Gordon et al., 2004). Complete excision of canine intracranial meningiomas may be difficult because of absence of a clear demarcation between affected and healthy brain tissue, tumour infiltration into healthy brain parenchyma and tumour friability. The median survival time of dogs with forebrain meningiomas treated surgically (standard surgery) ranges from 4.5 to 7 months (Kostolich and Dulisch, 1987; Axlund et al., 2002). The completeness of tumour gross resection has a positive impact on survival times. Surgical techniques that can improve the extent of tumour visualization and resection include intraoperative ultrasonography, endoscopy and use of an ultrasonic surgical aspirator. The latter enables tumour tissue ablation while sparing vasculature and minimizing damage to normal brain parenchyma (Gallagher et al., 1995; Greco et al., 2006; Klopp and Rao, 2009). The reported median survival time in dogs with meningioma removed with a surgical aspirator is 41.8 months
Structural Epilepsy

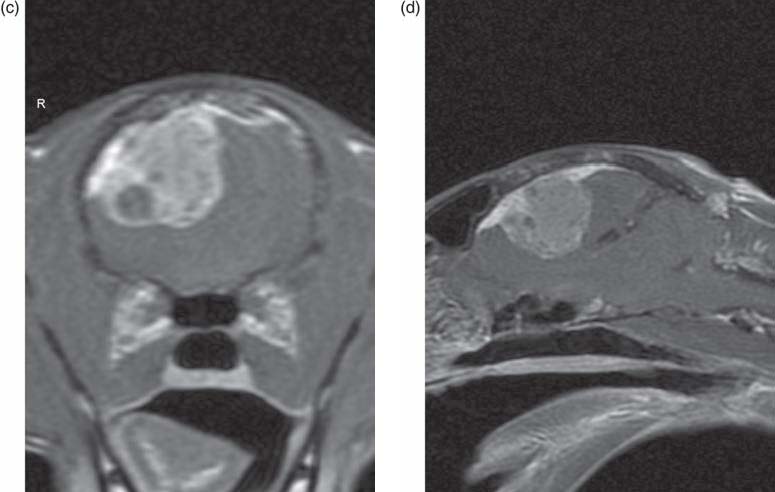
Fig. 5.18. MRI of the brain of a 13-year-old, female spayed, domestic short hair with a 2-week history of intermittent behavioural change, pacing and circling. The neurological examination indicated a right forebrain neuroanatomic diagnosis. Transverse T2W (a), T1W (b), T1WC (c) images at the level of the frontal lobe and dorsal (d) and paramedian T1WC (e) image show a well-defined rounded extra-axial mass in the right frontoparietal area. The lesion is moderately hyperintense on T2W (a), predominantly isointense in T1W (b) and heterogeneously contrast-enhancing on T1WC (c, d, e) with areas of no enhancement. The overlying and adjacent meninges are thickened and contrast-enhancing. The cerebral parenchyma is severely compressed and there is midline shift. The primary differential diagnosis for the extra-axial mass was meningioma.
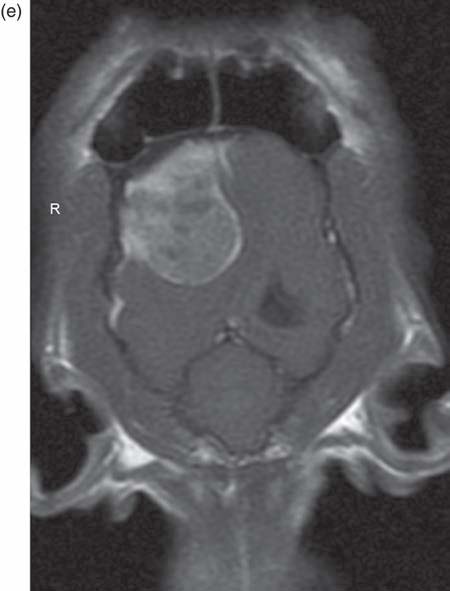
(Greco et al., 2006). Endoscopic assisted tumour resection resulted in a median survival time of
70.1 months in dogs with tumours (mostly meningiomas) located rostral to the tentorium cerebelli and of 23.4 months in dogs with tumours located caudal to the tentorium cerebelli (Klopp and Rao, 2009).
The median survival time of dogs with intracranial meningiomas treated with standard surgery followed by radiation therapy ranges from 15.7 to 30 months (Brearley et al., 1999; Théon et al., 2000; Axlund et al., 2002; Uriarte et al., 2011), which is longer than the survival reported with standard surgery alone. Proliferating cell nuclear antigen staining and extent and intensity of vascular endothelial growth factor expression can help to predict survival times in dogs treated with surgery and radiation for cerebral meningiomas (Théon et al., 2000; Platt et al., 2006). Information on survival times following radiation therapy of feline meningiomas is limited, probably because a successful outcome is frequently achieved with complete surgical removal. However, postoperative recurrence of feline meningiomas has been reported to occur in about 20% of cases (Gallagher et al., 1993; Gordon et al., 1994; Troxel et al., 2003) and a large study has reported a median time to tumour recurrence of 9.5 months after surgery (Troxel et al., 2003). Repeated surgery and/or radiotherapy may be successful in treating meningioma recurrence in cats.
The reported median survival time of dogs treated with radiotherapy and pharmacological palliative treatment for probable or confirmed meningiomas ranges from 12.2 to 20 months (Brearley, 1999; Bley, 2005). One cat with a suprasellar meningioma diagnosed on MRI was treated with orthovoltage radiation and lived for 8 months before dying of an unrelated neoplasm (Troxel et al., 2003).
There are limited reports on the use of chemotherapy for canine and feline meningiomas.
Hydroxyurea (30 to 50 mg/kg 3 days a week) has been used in the treatment of meningioma in dogs (Greco et al., 2006; Tamura et al., 2007). Hydroxyurea inhibits DNA synthesis without interfering with RNA or protein synthesis, and cell death occurs in the S phase of the cell cycle.
Structural Epilepsy
Hydroxyurea can cause progressive myelosuppression and therefore haematology should be monitored periodically. The survival time of a dog treated with dexamethasone and hydroxyurea was 14 months (Tamura et al., 2007). The clinical signs of this dog improved after 1 week of treatment, and the tumour size appeared reduced on MRI performed 37 days after treatment initiation (Tamura et al., 2007). The median survival time of 33 dogs with a presumptive MRI diagnosis of meningioma treated with glucocorticoids and hydroxyurea was 7 months (Cautela et al., 2009). The survival times of two dogs treated with surgery performed with a surgical aspirator and hydroxyurea were 33.3 and 50.8 months, respectively, with both dogs still alive at the time the study was concluded (Greco et al., 2006). The median survival time in five cats with tentorial meningiomas treated with surgery and hydroxyurea (20 mg/kg/day) was 20 months. No hydroxyurea-associated complications were observed (Forterre et al., 2006).
Lomustine (50 to 80 mg/m2 of body surface area at intervals of 6 to 8 weeks) also has been used to treat canine primary brain tumours (Fulton and Steinberg, 1990; Jung et al., 2006). Lomustine (CCNU) is an alkylating agent belonging to the nitroso-urea group. The high lipid solubility of lomustine and its metabolites results in wide distribution to tissues and penetration across the blood-brain barrier. Lomustine can cause myelosuppression (neutropaenia, anaemia and/or thrombocytopaenia), gastrointestinal (vomiting, diarrhoea and anorexia) and renal toxicity. Delayed dose-dependent hepatotoxicity occurs uncommonly (Heading et al., 2011). Haematology should be checked before treatment initiation and 1–2 weeks after each treatment course. Serum biochemistry should be monitored periodically. Lomustine is metabolized by hepatic microsomal (P450) enzymes, which are induced by phenobarbitone. Therefore these medications should not be used concurrently in the same animal to minimize the risk of reduced efficacy of lomustine. An alternative AEM to phenobarbitone such as levetiracetam or zonisamide should be administered to seizuring patients receiving lomustine. The survival time of a dog with an intracranial meningioma treated with prednisolone and lomustine was 13 months (Jung et al., 2006).
Pathological findings and classification
Grossly, canine and feline meningiomas are discrete, firm, rubbery, lobular, grey to pink, extramedullary masses. Sometimes they can be cystic or mineralized. Feline meningiomas are typically well encapsulated and displace the brain without invading it (Plate 16). The margins of canine meningiomas can be hard to distinguish from oedematous brain tissue. Nearly 30% of canine meningiomas invade normal brain tissue. Meningiomas can be attached to the dura in a broad, pedunculated, or sheet-like fashion (meningioma en plaque). Hyperostosis, or proliferation of the overlying skull in response to pressure from the meningioma, is seen mostly in cats (Summers et al., 2005; Vite, 2005). On histological examination, meningiomas are characterized by: (i) a mixture of sheets of epithelioid cells showing abundant and homogeneous cytoplasm without defined borders; this lobular pattern is characterized by syncytial whorl-like formations (meningothelial pattern) (Plate 15); and (ii) a number of spindle-shaped cells that create intersecting bundles or streams separated by variably dense collagen fibres (fibroblastic pattern) (Plate 16). Not infrequently, meningiomas have a mixture of meningothelial and fibroblastic patterns (transitional meningioma) or they may show a whorl formation with a core of central hyalinization, necrosis and mineralization (psammomatous pattern) (Motta et al., 2012). Angiomatous, papillary, granular, myxoid, microcystic and anaplastic subtypes have also been described. A histological classification scheme for grading canine meningiomas (based on the human World Health Organization (WHO) international classification scheme) has been proposed for canine meningiomas and includes: Grade I, benign; Grade II, atypical; and Grade III, malignant. Of 112 canine meningiomas, 56% were classified as Grade I, 43% were classified as Grade II and <1% were classified as Grade III (Sturges et al., 2008). Most intracranial meningiomas in cats are benign. In veterinary medicine, the value of such grading, from a prognostic and therapeutic standpoint, is not yet clear.
Extracranial metastases of meningiomas have been reported rarely in dogs and cats, occurring in the lungs and/or heart in dogs (Geib, 1966; Schulman et al., 1992; Dugan et al., 1993; Pérez et al., 2005) and in the kidneys and uterus in a cat (Dahme, 1957).
Astrocytoma
Astrocytic tumours are made up of neoplastic cells that phenotypically resemble astrocytes. The cell of origin of these tumours is still a matter of controversy (Stoica et al., 2011). Astrocytomas most frequently are located supratentorially in the frontal, temporal, parietal, pyriform lobes and olfactory bulb of the cerebrum and, less commonly, in the caudal fossa (Troxel et al., 2003; Young et al., 2011).
Incidence
Astrocytomas are the most common of the intra-axial primary CNS neoplasias in dogs and cats (Troxel et al., 2003; Snyder et al., 2006). Their frequency has been reported as 2.8% in cats and 17% in dogs with primary intracranial neoplasia (Troxel et al., 2003; Snyder et al., 2006). Boxers and some other brachycephalic breeds develop astrocytomas with higher frequency than other canine breeds (Snyder et al., 2006). Older dogs are most frequently affected, but astrocytomas can also occur in young animals (Kube et al., 2003; Walmsley et al., 2009; Wong et al., 2011). No gender over-representation has been reported. Astrocytomas typically appear as single lesions.
Clinical signs
Clinical signs commonly reflect the anatomic location and the secondary effects of the tumour. In one study in dogs, mentation change (12/25) and seizures (9/25 cases) were the most common presenting clinical signs (Snyder et al., 2006). The most common clinical signs of cats with astrocytomas were circling, seizures and altered mental status (Troxel et al., 2003).
Diagnostic investigations
Diagnostic investigations have been described in the general section on intracranial neoplasia and briefly include haematology, serum biochemistry, urinalysis, thoracic radiography, abdominal ultrasonography, brain MRI or CT, cerebrospinal fluid analysis and histology following biopsy or surgical removal.
Treatment and survival times
Treatment of intracranial astrocytomas can be divided into palliative (prednisolone and AEMs in seizuring animals) and definitive (surgery, radiation therapy, chemotherapy) (see general section on treatment of intracranial neoplasia). The median survival time of dogs with gliomas (astrocytomas and oligodendrogliomas) that underwent surgery followed by radiation therapy is 10.9 months (de Stefani et al., 2009). One cat with an astrocytoma treated with surgery and radiation therapy survived nearly 6 months (Troxel et al., 2003). Survival time for a cat with astrocytoma treated with corticosteroids alone was 35 days (Troxel et al., 2003).
Pathological findings and classification
The gross appearance of astrocytomas reflects their cellular composition, degree of differentiation and presence or absence of haemorrhage and necrosis (Summers et al., 2005). The least malignant astrocytomas present as greyish-white diffusely infiltrative tumours whose margins are difficult to define from normal neural tissue (Plate 19). The affected hemisphere may appear swollen. Anaplastic astrocytomas are more clearly delineated and often mottled reddish yellow with areas of haemorrhage and necrosis (Summers et al., 2005). Histological appearance varies. The human WHO 2007 histological classification scheme grades astrocytomas into four grades. Grades I and II astrocytomas (diffuse astrocytomas) are considered the least malignant forms and consist of a uniform, well-differentiated infiltrative cell population without mitotic activity. Grade III (anaplastic) astrocytomas have more nuclear atypia, a much higher cell density and mitotic activity. Grade IV astrocytomas (glioblastoma multiforme) are the most malignant and infiltrative frequently having regions of necrosis, micro-vascular proliferation, and sometimes intratumoural haemorrhage (Louis et al., 2007).
Structural Epilepsy


Fig. 5.19. MRI of the brain of a 5-year 6-month-old, male neutered boxer with acute onset of cluster seizures. The neuroanatomic localization was to the left forebrain. Transverse T2W (a), T1W (b) and T1WC (c), dorsal T1WC (d) and sagittal T2W (e) images show a large intra-axial ovoid mass in the ventral part of the left frontal lobe extending into the left ventral temporal lobe and mass effect. The mass lesion is heterogeneously hyperintense in T2W (a, e) and hypointense with scattered areas of hyperintensity in T1W (b) with peripheral ring-enhancement and patchy central enhancement on T1WC (c, d). The sagittal T2W image (e) shows frontal lobe hyperintensity (probable oedema) rostrally to the mass and severe subtentorial and foramen magnum herniations. The post-mortem histological diagnosis was oligodendroglioma.

Oligodendroglioma
Oligodendrogliomas appear to arise from the oligodendrocytic population. Oligodendrogliomas most frequently arise supratentorially in the frontal, pyriform (Fig. 5.19) and temporal lobes of the cerebrum and, less commonly, more caudally (Troxel et al., 2003; Young et al., 2011). These tumours can contact the meninges or the ventricles and sometimes break through the ependyma. Occasionally oligodendrogliomas can spread along CSF pathways to multiple sites within the brain and spinal cord (Koch et al., 2011). Most oligodendrogliomas grow by infiltration and destroy invaded tissue. The propensity for gliomas to abut the ventricular system and involve the pyriform lobe may be due to the potential mechanism of oncogenesis. Both the subventricular white matter and dentate gyrus, which closely approximates the pyriform lobe, contain neural stem cells, which may differentiate into gliomas (Young et al., 2011).
Incidence
The frequency of oligodendrogliomas has been reported as 2.6% in cats and 14% in dogs with primary intracranial neoplasia (Troxel et al., 2003; Snyder et al., 2006). Dogs with oligodendrogliomas are commonly middle aged or older (Snyder, et al., 2006; Young et al., 2011); however, oligodendrogliomas have been reported in dogs as young as 15 months (Triolo et al., 1994). As for astrocytomas, brachycephalic breeds such as boxers, Boston terriers and bulldogs are overrepresented (Snyder et al., 2006; Young et al., 2011). No gender over-representation has been reported (Snyder et al., 2006; Young et al., 2011).
Clinical signs
Clinical signs commonly reflect the anatomic location and the secondary effects of the tumour. In one study in dogs, seizures (18/25 cases) and mentation change (10/25) were the most common presenting clinical signs (Snyder et al.,
Structural Epilepsy
2006). Seizures were the most common clinical sign in cats with intracranial oligodendroglioma (Troxel et al., 2003).
Diagnostic investigations
Diagnostic investigations have been described in the general section on intracranial neoplasia and briefly include haematology, serum biochemistry, urinalysis, thoracic radiography, abdominal ultrasonography, brain MRI or CT, cerebrospinal fluid analysis and histology following biopsy or surgical removal.
Treatment and survival times
Treatment of intracranial oligodendrogliomas can be divided into palliative (prednisolone and AEMs in seizuring animals) and definitive (surgery, radiation therapy, chemotherapy) (see general section on treatment of intracranial neoplasia). The median survival time of dogs with gliomas (astrocytomas and oligodendrogliomas) that underwent surgery followed by radiation therapy is 10.9 months (de Stefani et al., 2009).
Pathological findings and classification
Grossly, oligodendrogliomas are usually pinkish to greyish, well-demarcated, gelatinous and soft, with multifocal haemorrhages and cystic areas. Tumour margins may be relatively sharp or undefined from normal neuroparenchyma. Focal extension into the overlying leptomeninges or into a ventricle commonly occurs (Summers et al., 2005). Histologically well-differentiated oligodendrogliomas have a very characteristic honeycomb or fried egg appearance due to artefactual cellular swelling producing perinuclear halos (Plate 20). Canine oligodendrogliomas are considered either low grade (Grades I or II) or high-grade (Grade III), with Grade III tumours greatly predominating.
CNS lymphoma
Intracranial lymphoma may be primary or metastatic within the CNS and either of the B- or T-cell type.
In dogs and cats CNS lymphoma occurs most commonly as part of multicentric disease (Troxel et al., 2003; Snyder et al., 2006). The forebrain as well as the brainstem, cerebellum and meninges can be affected.
Intravascular (angiotrophic) lymphoma is a rare disorder, with a predilection for the CNS that is characterized by neoplastic proliferation of malignant lymphoid cells within the lumen of blood vessels with little to no extension into adjacent parenchyma (McDonough et al., 2002).
Incidence
Lymphoma is the most common secondary intracranial tumour in cats and the third most common secondary intracranial tumour in dogs, after hemangiosarcoma and pituitary tumours (Troxel et al., 2003; Snyder et al., 2008). In one large case study, the rottweiler breed was over-represented in cases of CNS lymphoma and the mean age at diagnosis was 7.4 years (Snyder et al., 2008). Most cats with lymphoma are domestic short haired. The median age at diagnosis varies among studies ranging from 7 to 10.5 years of age in cats (Noonan et al., 1997; Troxel et al., 2003). Few cats test positive for feline leukaemia virus (Troxel et al., 2003). No gender over-representation has been reported (Troxel et al., 2003; Snyder et al., 2008).
Clinical signs
Clinical signs commonly reflect the anatomic location and the secondary effects of the tumour. Clinical signs are usually present for fewer than 30 days before presentation (Troxel et al., 2003; Snyder et al., 2008). The most commonly reported clinical signs in dogs include seizure, vestibular deficits and altered mental status (Snyder et al., 2008). In some affected dogs, no neurologic signs are detected despite CNS involvement (Snyder et al., 2008). The most common clinical signs reported in cats include anorexia, ataxia, lethargy, altered mental status and aggression (Troxel et al., 2003).
Diagnostic investigations
Diagnostic investigations have been described in the general section on intracranial neoplasia
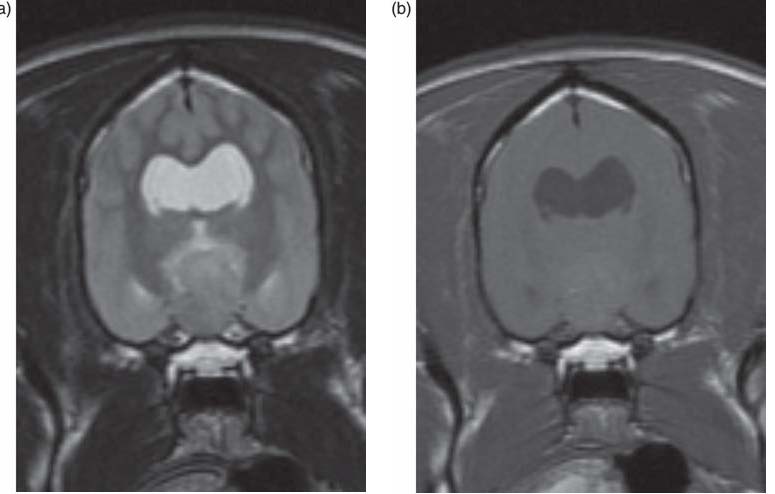
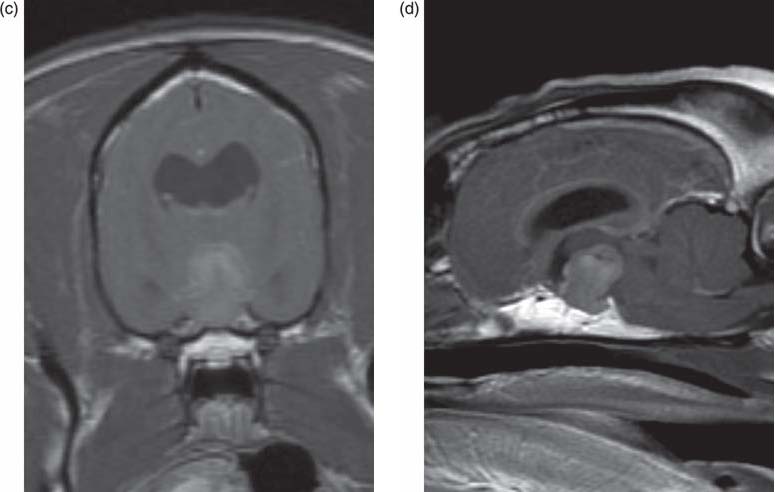
Fig. 5.20. MRI of the brain of a 9-year-old, female boxer with hyperadrenocorticism and recent onset of forebrain signs. Transverse T2W (a), T1W (b) and T1WC (c), and sagittal T1WC (d) images show an extra-axial mass in the middle cranial fossa, which appears iso- to mildly hyperintense in T2W (a), isointense in T1W (b) and moderately contrast enhances on T1WC (c, d). The mass completely effaces the pituitary gland and extends dorsally to the sella turcica compressing the interthalamic adhesion and rostral midbrain, resulting in obstructive hydrocephalus of the lateral ventricles. The main differential diagnosis for the middle cranial fossa mass was pituitary macroadenoma.
Structural Epilepsy
and briefly include haematology, serum biochemistry, urinalysis, thoracic radiography, abdominal ultrasonography, brain MRI or CT, cerebrospinal fluid analysis and histology following biopsy or surgical removal. Diagnosis of CNS lymphoma is supported by the identification of lymphoblasts in CSF (Plate 14), however their absence does not rule out lymphoma.
Treatment and survival times
CNS lymphoma can be treated with multi-drug protocols often including chemotherapeutic agents that cross the blood-brain barrier such as lomustine, cytosine arabinoside and temozolomide. Radiation therapy has also been used to treat CNS lymphoma. Survival times in dogs have not been well established. In two studies of cats treated with corticosteroids alone, the median survival was 21 and 35 days (Noonan et al., 1997; Troxel et al., 2003). In cats treated with chemotherapy and radiation for CNS lymphoma, median survival was 125 days (Noonan et al., 1997).
Pituitary tumours
Pituitary tumours usually arise from the adenohypophysis; tumours of the neurohypophysis are very rare. Pituitary tumours may cause signs of endocrine dysfunction, neurologic deficits or both. Neurologic signs occur as a consequence of compression or invasion of the tumour dorsally into the hypothalamus and diencephalon. Based on size, pituitary tumours can be divided into microtumours and macrotumours. Tumours larger than 10 mm in height or those extending dorsally above the sella turcica are considered macrotumours (Fig. 5.20a–d).
Incidence
Pituitary tumours are the second most common secondary tumour in dogs and cats (Snyder et al., 1998; Troxel et al., 2003). No breed and gender predilection has been reported in dogs, while a male predilection has been observed in cats (Elliott et al., 2000). Approximately 50% of dogs with pituitary macrotumours will develop neurologic signs secondary to the space-occupying effect of the mass, regardless of whether the dog has concurrent pituitary dependent hyperadrenocorticism (PDH) (Hanson et al., 2007).
Clinical signs
Clinical signs are related to the secretory nature of the tumour, the space-occupying effects of the mass, or both. Neurologic abnormalities attributed to a space-occupying pituitary tumour include change in behaviour, inappetence, obtundation, aggressiveness, anisocoria, apparent blindness and seizures. Depending on the tumour extent, additional neurologic signs include circling, ataxia, cranial nerve deficits involving vestibular, facial, trigeminal and oculomotor nerves. Endocrine abnormalities in dogs include PDH, and less commonly central diabetes insipidus or diabetes mellitus. Endocrine abnormalities in cats include insulin-resistant diabetes mellitus secondary to acromegaly (hypersomatotropism), and less commonly PDH.
Diagnostic investigations
Animals with intracranial neurologic signs as well as clinical signs suggestive of an endocrine disorder should undergo appropriate endocrine testing in addition to the investigations described in the general section on intracranial neoplasia (haematology, serum biochemistry, urinalysis, imaging of the chest, abdomen and brain). In dogs, testing of the pituitary adrenal axis should include low-dose dexamethasone suppression test, ACTH stimulation, urinary cortisol to creatinine ratio and abdominal ultrasound. In cats with suspect acromegaly, quantification of feline growth hormone may be performed. As with other intracranial tumours, MRI is the imaging modality of choice to investigate the pituitary gland, however a diagnosis can often be reached also with contrast-enhanced CT. The MRI features of pituitary tumours are described in Table 5.16. With CT, pituitary tumours typically demonstrate similar characteristics and patterns of contrast enhancement as observed with MRI. Dynamic imaging (MRI or CT) in which rapid imaging of the pituitary is performed during IV contrast administration can be used in the diagnosis of pituitary tumours and may increase the sensitivity of identifying microadenomas (Van der Vlugt-Meijer et al., 2007). The main differential diagnoses for a tumour located within the sellar and suprasellar region include meningioma, lymphoma, ependymoma, granular cell, germ cell tumours and craniopharyngiomas.
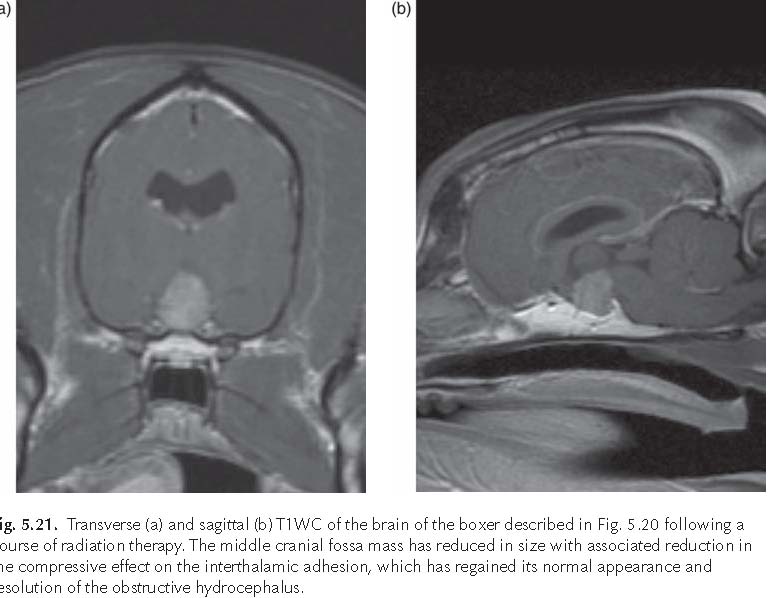
Treatment and survival times
Radiation therapy is the most commonly employed treatment for pituitary tumours in dogs and cats. Most studies evaluating radiation therapy for pituitary tumours demonstrated the benefits of decreasing tumour size, improving neurologic signs and prolonging survival (Fig. 5.21a, b). Median survival times in dogs vary among studies ranging from
11.8 months to over 43.6 months (Théon and Feldman, 1998; Bley et al., 2005; Kent et al., 2007). Presence and severity of neurologic and endocrine signs, variation in tumour biology and size as well as radiation therapy protocols may account for these differences in survival times. In one study, the median survival time of dogs with pituitary tumours treated with radiation therapy was not reached because dogs lived longer than the study period, while the median survival time of untreated dogs was nearly 12 months (Kent et al., 2007). Mean survival time in dogs with pituitary masses treated with radiation therapy was
46.8 months with 1-, 2- and 3-year estimated survival of 93, 87 and 55%, respectively. Mean survival time in untreated dogs was 18 months with 1-, 2- and 3-year estimated survival of 45, 32 and 25%, respectively. Dogs undergoing radiation therapy with smaller pituitary tumours lived longer than those with larger tumours (Kent et al., 2007). The pituitary-tobrain area and ratio of pituitary to-brain height can be used as prognostic factors for survival in dogs with pituitary tumours (Kent et al., 2007). In another study, median survival time of dogs with pituitary tumours having undergone radiation therapy was 43.6 months with all deaths attributed to disease progression or radiation damage (dogs that had died of other
Structural Epilepsy
causes or that were still alive at the time of data evaluation were censored) (Bley et al., 2005). Many dogs with PDH continue to require medical therapy for hyperadrenocorticism after radiation therapy (Théon and Feldman, 1998; Kent et al., 2007).
The median survival time of cats with pituitary tumours causing acromegaly and secondary diabetes mellitus without concurrent neurologic signs has been reported between 18 and 28 months (Brearley et al., 2006; Dunning et al., 2009). All cats had significant reduction in insulin requirements with approximately 50% of cats having resolution of diabetes mellitus allowing discontinuation of insulin therapy. Data on cats with neurologic signs secondary to pituitary tumours undergoing radiation therapy are limited. Overall response to treatment appears favourable with survival times ranging from 1 to 66 months (Kaser-Hotz et al., 2002; Brearley et al., 2006).
Microsurgical trans-sphenoidal hypophysectomy has been reported to be relatively safe and effective as the sole treatment for pituitary tumours in dogs and cats (Meij et al., 2002). Pituitary surgery requires a team approach and the neurosurgeon performing hypophysectomies must master a learning curve. Old age, large pituitary size, and high preoperative concentrations of plasma adrenocorticotropic hormone were associated with an increased risk of PDH-related death in dogs with PDH (Hanson et al., 2007). Survival and risk of recurrence can be influenced by age, pituitary size, endocrine factors and thickness of the sphenoid bone. Postoperatively, dogs must be supplemented with corticosteroids, thyroxine and vasopressin to prevent crises related to hypoadrenocorticism, hypothyroidism and diabetes insipidus, respectively.
Degenerative
Degenerative diseases of the CNS include lysosomal storage diseases, organic acidurias, mitochondrial encephalopathies, leukodystrophies, spongy degenerations and multisystem neuronal degenerations/abiotrophies.
This section will focus on degenerative disorders that have been reported to cause seizures.
Lysosomal storage diseases
Lysosomal storage diseases are rare degenerative disorders that result from a genetically determined defect of a specific lysosomal acid hydrolase enzyme. This results in accumulation and storage of metabolic products within the cytoplasm of neurons and/or glia throughout the nervous system, as well as in cells in other organs (Braund, 2003). Most lysosomal storage diseases have been recognized in specific breeds of dogs or cats, have an autosomal recessive mode of inheritance, and affect both males and females. Animals are usually normal at birth, but they generally fail to grow normally. Clinical signs occur within the first several weeks to several months of life in most storage disorders. However, onset of signs may be delayed until adulthood with some conditions such as some of the glycoproteinoses and neuronal ceroid lipofuscinoses. Most lysosomal storage diseases, except for the mucopolysaccharidoses, exhibit involvement of the nervous system and/or other organs including the eye. Skeletal abnormalities and craniofacial malformations are prominent in the mucopolysaccharidoses and a common feature in mannosidosis and mucolipidosis II. Enlargement of visceral organs
(e.g. hepatomegaly, splenomegaly) is observed with some glycogenoses, sphingomyelinoses and mannosidoses. Neurologic signs vary depending on the specific enzymatic defect and reflect involvement of the cerebellum, forebrain, spinal cord and sometimes peripheral nervous system (PNS). Cerebellar dysfunction is the first sign of several storage diseases including mannosidosis, gangliosidosis, galactosialidosis, glucocerebrosidosis, globoid cell leukodystrophy sphingomyelinosis and some forms of neuronal ceroid lipofuscinoses. Forebrain dysfunction including seizures predominates in some forms of neuronal ceroid lipofuscinosis and in glycoproteinoses (Lafora disease and fucosidosis). The lysosomal storage disorders reported to cause seizures in dogs and cats are listed in Table 5.17.
Table 5.17. Lysosomal storage disorders reported to cause seizures in dogs and cats (modified from Lorenz et al., 2011, 5th edn. Handbook of Veterinary Neurology, Saunders-Elsevier. Systemic or multifocal signs, Chapter 15, pp. 434–437).
| Disease subgroup | Lysosomal storage disease | Enymatic deficiency | Affected species or breed | Age at onset | Clinical signs | Diagnosis |
| Glycoproteinoses | Lafora disease | α-Glucosidase | Beagle, Basset hound, Poodle, | 5–9 m 3 y | Myoclonic seizures that can be precipitated by | |
| Wirehaired miniature dachshund, DSH | 9–12 y 5–8 y | auditory or visual stimuli, obtundation | ||||
| Fucosidosis | α-L-Fucosidase | English springer spaniel | 6 m–3 y | Cerebellar ataxia, behavioural abnormalities, visual and hearing deficits, dysphonia, | DNA testing | |
| dysphagia, seizures; enlarged peripheral (ulnar) nerves | ||||||
| Oligosaccharidoses Sphingolipidoses | Glycogenosis type 1 GM1-gangliosidosis type 1 | Glucose-6phosphatase β-D-Galactosidase | Silky terrier, Maltese, other toy breeds, DSH Beagle cross, Portuguese water dog, English | Weeks 4–7 m 2–3 m | Weakness, seizures, stupor Cerebellar ataxia, corneal clouding, tremor, seizures, paralysis, skeletal, facial | |
| springer spaniel, Alaskan husky, Shiba Inu dog, | dysmorphism | |||||
| DSH | ||||||
| Metachromatic leukodystrophy | Arylsulfatase A | DSH | 2 w | Progressive motor dysfunction, seizures, | ||
| opisthotonus, neuropathy |
Muscle biopsy, DNA testing (dachshund)
Urine screening
DNA testing (Alaskan husky, Shiba dog)
188 L. De Risio
| Proteinoses ceroid | CLN 1 | Palmitoyl protein | Dachshund | Months | Behaviour abnormalities, | Tissue biopsy |
| lipofuscinoses | thioesterase I | visual deficits, cerebellar | (autofluorescence), | |||
| (CLN) | CLN 2 | Tripeptidyl-peptidase | Dachshund | 4–5 m | ataxia, myoclonus, | DNA testing for |
| CLN 4 | Arylsulfatase G | American | >1 y | seizures | CLN 2, CLN 4, | |
| Staffordshire | CLN 5, CLN 6, | |||||
| terrier | CLN 8, Cathepsin D | |||||
| CLN 5 | Soluble lysosomal | Border collie | 2 y | (see Table 10.6) | ||
| membrane protein | ||||||
| CLN 6 | Endoplasmic | Australian | 1–2 y | |||
| reticulum | shepherd | |||||
| membrane protein | ||||||
| CLN 8 | Membrane protein of | English setter | 2 y | |||
| the endoplasmic | ||||||
| reticulum | ||||||
| Cathepsin D | Cathepsin D | American bulldog | 2–4 y | |||
| Unknown | Unknown | Several canine | m–y | |||
| breeds, DSH, | ||||||
| Siamese cat |
w = weeks; m = months; y = years; DSH, Domestic short hair
Structural Epilepsy
PNS involvement can occur with fucosidosis, sphingomyelinosis type A, mannosidosis, glycogenosis type IV and globoid cell leukodystrophy. Myopathy (muscle weakness, exercise intolerance) is a prominent feature of the glycogenoses, but forebrain signs may result from hypoglycaemia. Lysosomal storage disorders are progressive (most commonly slowly, but sometimes also rapidly) and eventually fatal.
A presumptive diagnosis is based on signalment, clinical signs and exclusion of other diseases. Vacuolated leukocytes may be detected on blood, bone marrow or CSF. Metabolic by-products can be identified in the urine by specific assays in certain storage disorders including glycogenosis type 1, mannosidosis and mucopolysaccaridosis. Muscle and/or nerve biopsy may support the diagnosis of some lysosomal storage diseases (e.g. those with PNS involvement, Lafora disease). The MRI features of ceroid lipofuscinosis, globoid cell leukodystrophy and GM1- and GM2gangliosidosis have been reviewed (Hecht and Adams, 2010b). Definitive diagnosis of a specific lysosomal storage disease can be reached by genetic testing for known mutations in blood, identifying the deficient enzyme activity on tissue biopsies (e.g. liver or cultured fibroblasts) or blood leukocytes, or identification of the storage product (generally post-mortem).
There is no treatment for these diseases; however, experimental therapeutic approaches including enzyme replacement, cell transplantation and gene therapy are under investigation.
A comprehensive review of all storage disorders is beyond the scope of this text and the reader should refer to the references for additional information (Skelly and Franklin, 2002; Braund, 2003).
Organic acidurias
Organic acidurias are characterized by an error of cellular metabolism leading to the accumulation of one or more organic acids, which are often detectable in the serum, CSF or urine by gas chromatography-mass spectroscopy (Sewell et al., 2007). The metabolic defect may be due to the lack of an essential enzyme or inadequate function of an enzyme due to insufficiency of a cofactor (e.g. deficiency of cobalamin (vitamin B12) will cause dysfunction of methylmalonic CoA decarboxylase in the Kreb’s cycle). Mitochondrial respiratory chain enzymes or cytosolic enzymes can be affected. The majority of organic acidurias are inherited, usually with an autosomal recessive pattern; however, they can also be acquired secondary to a malabsorption (e.g. exocrine pancreatic insufficiency) or toxicosis (e.g. propylene glycol). Clinical signs of encephalopathy can result from abnormal cellular metabolism, toxic effects of the accumulated organic acid, or both. In addition, the error in the cellular metabolism can lead to other metabolic abnormalities such as hypoglycaemia, lactic acidosis, ketoacidosis, or hyperammonaemia, which may contribute to cause encepahlopathic signs. Reported organic acidurias in dogs and cats include L-2-hydroxyglutaric aciduria in Staffordshire bull terriers, Yorkshire terriers and a West Highland white terrier, malonic aciduria in Maltese dogs, methylmalonic aciduria and D-lactic acidosis in cats.
Staffordshire bull terriers with L-2hydroxyglutaric aciduria develop neurologic signs between 4 months and 7 years of age. Four of six dogs had chronic progressive disease, and two dogs presented with an acute onset of seizures between 4 and 6 months of age (Abramson et al., 2003).
Neurologic signs included seizures, ataxia, dementia and head tremor. Levels of L-2hydroxyglutaric acid and lysine were elevated in urine, plasma and CSF. MRI revealed bilaterally symmetric, diffuse regions of grey matter (mostly in the thalamus, hypothalamus, dentate nucleus, basal ganglia, dorsal brainstem, cerebellar nuclei and cerebellar gyri), which appeared hyperintense on T2-W images, hypointense in T1-W images, did not contrast enhance and did not exhibit a mass effect (Fig. 5.22a–e). CSF cytology and protein concentration were normal. A mutation in the dehydrogenase that metabolizes L-2-hydroxyglutaric acid has been described (Penderis et al., 2007) and a genetic test is available to diagnose this condition. No definitive treatment for the inborn error of metabolism is available. Phenobarbitone (3 mg/kg/day) has been reported to adequately control seizures in affected Staffordshire bull terriers (Abramson
Structural Epilepsy


Fig. 5.22. MRI of the brain of a 7-month-old Staffordshire bull terrier with a history of three episodes of suspected seizure activity and multifocal intracranial neuroanatomic localization. Transverse T2W (a), FLAIR (b), T1W (c), T1WC (d) images show bilaterally symmetrical signal changes in the grey matter of the thalamus and cerebral cortex, which are hyperintense on T2W (a), FLAIR (b), iso- to hypointense on T1W and do not contrast-enhance. The sagittal T2W (e) image shows grey matter hyperintensity at the level of the dorsal brainstem, cerebral and cerebellar cortex. L-2 hydroxyglutaric aciduria was confirmed with a genetic test.

et al., 2003). Cobalamin supplementation was implemented (25 mg/kg SC) in Staffordshire bull terriers with concurrent mild methylmalonic aciduria. One Staffordshire bull terrier with L-2-hydroxyglutaric aciduria and low muscle carnitine concentration was supplemented with L-carnitine (50 mg/kg PO q12 h), vitamin B1 (thiamine, 89 mg PO q24 h) and riboflavin (100 mg PO q24 h) (Abramson et al., 2003).
One West Highland white terrier with L-2-hydroxyglutaric aciduria presented at 5 years of age with a long-standing history of gait abnormality affecting all four limbs, impaired vision, behaviour abnormalities (loss of obedience training and staring at walls) and recent episodes of severe head tremor when stressed. The clinical signs had an insidious onset and progressively became more severe (Garosi et al., 2005). Neurologic examination revealed forebrain, brainstem and cerebellar signs. MRI findings were similar to those described in the Staffordshire bull terriers.
Levels of L-2-hydroxyglutaric acid were elevated in urine.
Two female Yorkshire terrier dogs with generalized tonic-clonic seizures, MRI findings similar to those described in the Staffordshire bull terriers and West Highland white terrier with L-2-hydroxyglutaric aciduria and elevated levels of L-2-hydroxyglutaric acid in the urine, had a homozygous mutation at the translation initiation codon of the L-2-hydroxyglutaric dehydrogenase gene (Sanchez- Masian et al., 2012). The same mutation has been identified in an 8-month-old Yorkshire terrier that presented with episodes of hyperactivity and aggressive behaviour (Farias et al., 2012).
Maltese dogs from a family with malonic aciduria developed seizures at 6 months of age and later progressed to stupor. Hypoglycaemia, acidosis and ketonuria were identified. The clinical signs resolved after the diet was altered to feed one that was high in carbohydrate and low in fat (O’Brien et al., 1999).
Structural Epilepsy
Cobalamin (vitamin B12) deficiency has been associated with methylmalonic aciduria and encephalopathic signs in young cats (Vaden et al., 1992; Kelmer et al., 2007). It was hypothesized that the cobalamin deficiency was due to deficiency of an intrinsic factor necessary for cobalamin absorption. Neurological and MRI findings consistent with a polioencephalopathy resolved following cobalamin supplementation in a cat with cobalamin deficiency of unknown cause (Simpson et al., 2012).
Suspected acquired D-lactic acidosis secondary to gastrointestinal disease has been reported in a 2-year-old cat with weight loss and polyphagia of 1 year duration and recent episodic weakness, ataxia, lethargy and obtundation. Bacterial overgrowth was considered the cause of the over-production of the D-isoform of lactic acid (Packer et al., 2005).
Mitochondrial encephalopathies and encephalomyelopathies
Mitochondrial encephalopathies and encephalomyelopathies result from various defects of mitochondrial respiratory enzyme function. There are similarities between the pathogenesis of mitochondrial encephalopathies and organic acidurias. The main difference is the absence or presence, respectively, of detectable organic acids in the serum, CSF or urine by gas chromatography-mass spectroscopy. Mitochondrial encephalopathies or encephalomyelopathies have been described in Australian cattle dogs (Brenner et al., 1997a), Alaskan huskies (Brenner et al., 2000), Shetland sheepdogs, Yorkshire terriers, English springer spaniels (Brenner et al., 1997b) and in cats. The majority of animals develop clinical signs between 4 and 15 months of age. However, earlier or later onset has been reported in a few dogs. Clinical signs include various combinations of the following: seizures, behavioural abnormalities, obtundation, propulsive pacing, generalized ataxia, tetraparesis, visual deficits, head tremor, facial hypalgesia and difficulties in prehension of food, dysphagia, dysphonia and tremors. Onset of signs can be acute
Clinical signs can be slowly progressive
(e.g. Australian cattle dogs, Shetland sheepdogs) or episodic (e.g. Alaskan huskies). MRI may reveal bilaterally symmetric cavitary lesions in the brain and/or spinal cord, which are isointense or hypointense on T1-W images, hyperintense on T2-W images, do not contrast-enhance and do not have a mass effect (Harkin et al., 1999). CSF analysis can be normal or reveal mild increase in protein and mild mononuclear pleocytosis. Elevated serum and CSF levels of lactate and pyruvate have been reported in the Australian cattle dogs. Definitive diagnosis is based on characteristic gross and histologic findings in the CNS post-mortem. Gross findings include bilateral and symmetrical soft grey cavitated foci primarily affecting the CNS grey matter (Plate 21). Lesion distribution varies among breeds. Histologically, lesions are characterized by neuronal loss, spongiosis, vascular hypertrophy and hyperplasia, gliosis and cavitation. Ultrastructurally mitochondrial abnormalities can be observed in neurons and astrocytes. To date, no effective treatment has been reported.
Leukodystrophies
Leukodystrophies are rare, probably inherited disorders of myelin synthesis and maintenance. They involve CNS myelin with a typically bilaterally symmetrical, often regional, distribution (Braund, 2003). Leukodystrophies have been reported in the Dalmatian, Labrador retriever, Scottish terrier, bull mastiff and miniature poodle (Braund, 2003; Morrison et al., 2006). Onset of neurologic signs is within 3 to 6 months of age. Clinical signs include UMN paraparesis, which progresses to tetraparesis and sometimes also seizures, abnormal behaviour and mental status, ataxia, generalized tremors and visual deficits. Clinical signs are progressive and eventually fatal. A presumptive diagnosis is based on signalment, clinical signs and exclusion of other diseases. Definitive diagnosis is based on post-mortem histologic findings of extensive CNS myelin degeneration and replacement by severe astrogliosis or Rosenthal fibres (astrocytic processes). There is no treatment (Braund, 2003).
Other myelin disorders, resulting predominantly or exclusively in spinal cord dysfunction, are not included in this text.
Spongy degenerations
The term spongy degeneration refers to neurodegenerative disorders characterized by vacuolation of the affected tissue. Vacuolation can occur with separation of the myelin sheath or within the neuronal cell body.
A spongiform leukoencephalomyelopathy has been reported in the Australian cattle dog, Shetland sheepdog and Labrador retriever. Clinical signs including cerebellar ataxia, seizures and opisthotonus occur within the first few weeks to months of life and are progressive.
Forebrain signs, including seizures and behavioural changes have been reported in young saluki dogs and cocker spaniels with spongy degeneration involving predominantly the grey matter.
As with other neurodegenerative CNS disorders, a presumptive diagnosis is based on signalment, clinical signs and exclusion of other diseases. Definitive diagnosis of spongiform leukoencephalomyelopathy is based on post-mortem histologic findings of widespread vacuolation and myelin degeneration of the spinal cord, brainstem, cerebellum and cerebral white matter. Definitive diagnosis of spongy degeneration predominantly in the CNS grey matter is based on post-mortem histology. There is no treatment (Braund, 2003).
Multisystem neuronal degeneration/abiotrophy
Abiotrophy is a process by which cells develop normally but later degenerate due to an intrinsic cellular defect. The degeneration primarily involves neuronal cell bodies in the cerebellum, cerebrum, brainstem, spinal cord, nerves or multiple systems. Clinical signs relate to the predominant region of the nervous system affected and are progressive. Most abiotrophies in dogs and cats are characterized by clinical signs primarily or exclusively related to cerebellar dysfunction (cerebellar ataxia and intention tremor). Forebrain and cerebellovestibular dysfunction have been reported in a group of related red-haired cocker spaniels with a multisystem neuronal abiotrophy. Clinical signs occurred within 10 to 14 months of age and included behaviour abnormalities, generalized-onset seizures, intention tremor, cerebellar ataxia, circling, vision loss and proprioceptive deficits. Progression is generally slow over several months and in most instances eventually fatal. Definitive diagnosis is based on histological evidence of widespread neuronal cell loss throughout the brain. There is no treatment (Jaggy and Vandevelde, 1988; Braund, 2003).
References
Abramson, C.J., Platt, S.R., Jakobs, C., Verhoeven, N.M., Dennis, R., Garosi, L. and Shelton, G.D. (2003) L-2Hydroxyglutaric aciduria in Staffordshire bull terriers. Journal of Veterinary Internal Medicine 17, 551–556.
Adamo, P.F., Adams, W.M. and Steinberg, H. (2007a) Granulomatous meningoencephalomyelitis in dogs. Compendium on Continuing Education for the Practicing Veterinarian 29(11), 678–690.
Adamo, P.F., Rylander, H. and Adams, W.M. (2007b) Cyclosporin use in multidrug therapy for meningoencephalomyelitis of unknown aetiology in dogs. Journal of Small Animal Practice 48(9), 486–496. Addie, D.D. (2012) Feline coronavirus infections. In: Infectious Diseases of the Dog and Cat, 4th edn.
Saunders-Elsevier, London, pp. 92–108. Agrawal, A., Timothy, J., Pandit, L. and Manju, M. (2006) Post-traumatic epilepsy: an overview. Clinical Neurology and Neurosurgery 108, 433–439.
Altay, U.M., Skerritt, G.C., Hilbe, M., Ehrensperger, F. and Steffen, F. (2011) Feline cerebrovascular disease: clinical and histopathologic findings in 16 cats. Journal of American Animal Hospital Association 47(2), 89–97.
Annegers, J.F., Hauser, W.A., Coan, S.P. and Rocca, W.A. (1998) A population-based study of seizures after traumatic brain injuries. The New England Journal of Medicine 338(1), 20–24.
Structural Epilepsy
Añor, S., Sturges, B.K., Lafranco, L., Jang, S.S., Higgins, R.J., Koblik, P.D. and LeCouteur, R.A. (2001) Systemic phaeohyphomycosis (Cladophialophora bantiana) in a dog–clinical diagnosis with stereotactic computed tomographic-guided brain biopsy. Journal of Veterinary Internal Medicine 15(3), 257–261.
Arntz, R., Rutten-Jacobs, L., Maaijwee, N., Schoonderwaldt, H., Dorresteijn, L., van Dijk, E. and de Leeuw, F.E. (2013) Post-stroke epilepsy in young adults: a long-term follow-up study. PLoS One 8(2), e55498. doi: 10.1371/journal.pone.0055498.
Axlund, T.W., McGlasson, M.L. and Smith, A.N. (2002) Surgery alone or in combination with radiation therapy for treatment of intracranial meningiomas in dogs: 31 cases (1989–2002). Journal of American Veterinary Medical Association 221, 1597–1600.
Bagley, R.S. (2003a) Intracranial surgery. In: Textbook of Small Animal Surgery. Saunders Elsevier, Philadelphia, Pennsylvania, pp. 1261–1277.
Bagley, R.S. (2003b) Surgical approaches to the central nervous system (brain). In: Textbook of Small Animal Surgery. Saunders Elsevier, Philadelphia, Pennsylvania, pp. 1163–1173.
Bagley, R.S., Gavin, P.R., Moore, M.P., Silver, G.M., Harrington, M.L. and Connors, R.L. (1999) Clinical signs associated with brain tumors in dogs: 97 cases (1992-1997). Journal of the American Veterinary Medical Association 215, 818–819.
Bailey, C.S. and Higgins. R.J. (1986) Characteristics of cisternal cerebrospinal fluid associated with primary brain tumors in the dog: a retrospective study. Journal of American Veterinary Medical Association 188, 414–417.
Barreau, P., Dunn, K. and Fourie, Y. (2010). Canine meningioma: A case report of a rare subtype and novel atlanto basioccipital surgical approach. Veterinary and Comparative Orthopaedics and Traumatology 23, 372–376.
Bateman, S.W. and Parent, J.M. (1999) Clinical findings, treatment, and outcome of dogs with status epilepticus or cluster seizures: 156 cases (1990–1995). Journal of American Veterinary Medical Association 215, 1463–1468.
Bathen-Noethen, A., Stein, V.M., Puff, C., Baumgaertner, W. and Tipold, A. (2008) Magnetic resonance imaging findings in acute canine distemper virus infection. Journal of Small Animal Practice 49(9), 460–467.
Bauer, J., Vezzani, A. and Bien, C.G. (2012) Epileptic encephalitis: the role of the innate and adaptive immune system. Brain Pathology 22(3), 412–421.
Belcastro, V., Pierguidi, L. and Tambasco, N. (2011) Levetiracetam in brain ischemia: clinical implications in neuroprotection and prevention of post-stroke epilepsy. Brain & Development 33(4), 289–293.
Bentley, R.T., Faissler, D. and Sutherland-Smith, J. (2011) Successful management of an intracranial phaeohyphomycotic fungal granuloma in a dog. Journal of American Veterinary Medical Association 15, 239(4), 480–485.
Biel, M., Kramer, M., Forterre, F., Jurina, K., Lautersack, O., Failing, K. and Schmidt, M.J. (2013) Outcome of ventriculoperitoneal shunt implantation for treatment of congenital internal hydrocephalus in dogs and cats: 36 cases (2001–2009). Journal of American Veterinary Medical Association 242, 948–958.
Bley, C.R., Sumova, A., Roos, M. and Kaser-Hotz, B. (2005) Irradiation of brain tumors in dogs with neurologic disease. Journal of Veterinary Internal Medicine 19, 849–854.
Bowen, A. and Greene, C.E. (2012) West Nile virus infection. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 214–217.
Bradley, W.G. (1993) MR appearance of hemorrhage in the brain. Radiology 189(1), 15–26.
Braund, K.G. (2003) Storage Disorders. In: Braund, K.G. (ed.) Clinical Neurology in Small Animals – Localization, Diagnosis and Treatment . International Veterinary Information Service (www.ivis.org), Ithaca, New York.
Brearley, M.J., Jeffery, N.D., Phillips, S.M. and Dennis, R. (1999) Hypofractionated radiation therapy of brain masses in dogs: a retrospective analysis of survival of 83 cases (1991–1996). Journal of Veterinary Internal Medicine 13, 408–412.
Brearley, M.J., Polton, G.A., Littler, R.M. and Niessen, S.J. (2006) Coarse fractionated radiation therapy for pituitary tumours in cats: a retrospective study of 12 cases. Journal of Veterinary Comparative Oncology 4, 209–217.
Brenner, O., de Lahunta, A., Summers, B.A., Cummings, J.F., Cooper, B.J., Valentine, B.A. and Bell, J.S. (1997a) Hereditary polioencephalomyelopathy of the Australian cattle dog. Acta Neuropathologica 94(1), 54–66.
Brenner, O., de Lahunta, A., Cummings, J.F., Summers, B.A. and Monachelli, M. (1997b) A canine encephalomyelopathy with morphological abnormalities in mitochondria. Acta Neuropathologica 94, 390–397.
Brenner, O., Wakshlag, J.J., Summers, B.A. and de Lahunta, A. (2000) Alaskan husky encephalopathy – a canine neurodegenerative disorder resembling subacute necrotizing encephalomyelopathy (Leigh syndrome). Acta Neuropathologica 100(1), 50–62.
Bromel, C. and Greene, C.E. (2012) Histoplasmosis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 614–621.
Buckmaster, P.S., Smith, M.O., Buckmaster, C.L., LeCouteur, R.A. and Dudek, F.E. (2002) Absence of Temporal lobe epilepsy pathology in dogs with medically intractable epilepsy. Journal of Veterinary Internal Medicine 16, 95–99.
Cautela, M.A., Dewey, C.W., Cerda-Gonzalez, S., Fletcher, D.J. and Barone, G. (2009) Oral hydroxyurea therapy for dogs with suspected intracranial meningioma: a retrospective cohort study (2004–2009). Journal of Veterinary Internal Medicine 23, 737.
Cervera, V., Mai, W., Vite, C.H., Johnson, V., Dayrell-Hart, B. and Seiler, G.S. (2011) Comparative magnetic resonance imaging findings between gliomas and presumed cerebrovascular accidents in dogs. Veterinary Radiology and Ultrasound 52(1), 33–40.
Chen, J.W., Ruff, R.L., Eavey, R. and Wasterlain, C.G. (2009) Posttraumatic epilepsy and treatment. Journal of Rehabilitation Research and Development 46(6), 685–696.
Cherubini, G.B., Rusbridge, C., Singh, B.P., Schoeniger S. and Mahoney, P. (2007) Rostral cerebellar arterial infarct in two cats. Journal of Feline Medicine and Surgery 9(3), 246–253.
Chesnut, R.M., Temkin, N., Carney, N., Dikmen, S., Rondina, C., Videtta, W., Petroni, G., Lujan, S., Pridgeon, J., Barber, J., Machamer, J., Chaddock, K., Celix, J.M., Cherner, M. and Hendrix, T. (2012) A trial of intracranial-pressure monitoring in traumatic brain injury. The New England Journal of Medicine 367(26), 2471–2481.
Christensen, J., Pedersen, M.G., Pedersen, C.B., Sidenius, P., Olsen, J. and Vestergaard, M. (2009) Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 28, 373(9669), 1105–1110.
Coates, J.R., Axlund, T.W., Dewey, C.W. and Smith, J. (2006) Hydrocephalus in Dogs and Cats. Compendium on Continuing Education for the Practicing Veterinarian 136–145.
Coates, J.R., Barone, G., Dewey, C.W., Vitale, C.L., Holloway-Azene, N.M. and Sessions, J.K. (2007) Procarbazine as adjunctive therapy for treatment of dogs with presumptive antemortem diagnosis of granulomatous meningoencephalomyelitis: 21 cases (1998–2004). Journal of Veterinary Internal Medicine 21, 100–106.
Cocayne, C.G. and Cohn, L.A. (2012) Ehrlichia ewingii infection (Canine granulocytotropic ehrlichiosis). In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 241–244.
Conrad, J., Pawlowski, M., Dogan, M., Kovac, S., Ritter, M.A. and Evers, S. (2013) Seizures after cerebrovascular events: Risk factors and clinical features. Seizure pii: S1059–1311(13)00028-9. doi: 10.1016/j. seizure.2013.01.014.
Cooley, A.J., Clemmons, R.M. and Gross, T.L. (1987) Heartworm disease manifested by encephalomyelitis and myositis in a dog. Journal of the American Veterinary Medical Association 190(4), 431–432.
Costanzo, C., Garosi, L.S., Glass, E.N., Rusbridge, C., Stalin, C.E. and Volk, H.A. (2011) Brain abscess in seven cats due to a bite wound: MRI findings, surgical management and outcome. Journal of Feline Medicine and Surgery 13(9), 672–680.
Crook, K.I., Early, P.J., Messenger, K.M., Muñana, K.R., Gallagher, R. and Papich, M.G. (2013) The pharmacokinetics of cytarabine in dogs when administered via subcutaneous and continuous intravenous infusion routes. Journal of Veterinary Pharmacology and Therapeutics 36(4), 408–411.
Dahme, E. (1957) Meningiome bei Fleischfressern. Berliner und Münchener Tierärztliche Wochenschrift 70, 32–34.
Davies, E.S., Volk, H.A., Behr, S., Summers, B., de Lahunta, A., Syme, H., Jull, P. and Garosi, L. (2012) Porencephaly and hydranencephaly in six dogs. The Veterinary Record 170(7), 179.
Day, M.J. (2012a) Canine disseminated aspergillosis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 662–666.
Day, M.J. (2012b) Feline disseminated aspergillosis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, p. 666.
Day, M.J. and Barrs, V.R.D. (2012) Feline sinonasal and sinoorbital Aspergillus fumigatus complex and Penicillum infections. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 659–662.
Day, M.J., Peeters, D. and Clercx, C. (2012) Canine sinonasal aspergillosis-penicillosis In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 651–659.
Structural Epilepsy
de Stefani, A., De Risio, L. and Matiasek, K. (2008) Intravenous cytosine arabinoside in the emergency treatment of 9 dogs with central nervous system inflammatory disease of unknown etiology. Journal of Veterinary Internal Medicine 22, 508.
de Stefani, A., Sparkes, A., Garosi, L.S., De Risio, L., Llabres, F.J. and Platt, S.R. (2009) Clinical signs, magnetic resonance imaging findings and survival in dogs with intracranial meningiomas and glial cell tumours. Journal of Veterinary Internal Medicine 23, 737.
de Stefani, A., de Risio, L., Platt, S.R., Matiasek, L., Lujan-Feliu-Pascual, A. and Garosi, L.S. (2011) Surgical technique, postoperative complications and outcome in 14 dogs treated for hydrocephalus by ventriculoperitoneal shunting. Veterinary Surgery 40(2), 183–191.
DeLahunta, A. and Glass, E. (2009a) Cerebrospinal fluid and hydrocephalus. In: Veterinary Neuroanatomy and Clinical Neurology. Saunders-Elsevier, St Louis, Missouri, pp. 54–76.
DeLahunta, A. and Glass, E. (2009b) Development of the nervous system: malformation. In: Veterinary Neuroanatomy and Clinical Neurology. Saunders-Elsevier, St Louis, Missouri, pp. 23–53.
Dennis, M.M., Pearce, L.K., Norrdin, R.W. and Ehrhart, E.J. (2005) Bacterial meningoencephalitis and ventriculitis due to migrating plant foreign bodies in three dogs. The Veterinary Pathology 42(6), 840–844.
Dewey, C.W. (2000) Emergency management of the head trauma patient. The Veterinary Clinics of North America Small Animal Practice 30, 207–225.
Diaz-Arrastia, R., Agostini, M.A., Madden, C.J. and Van Ness, P.C. (2009) Posttraumatic epilepsy: the endophenotypes of a human model of epileptogenesis. Epilepsia 50(Suppl. 2), 14–20.
DiBartola, S.P., Johnson, S.E., Johnson, G.C. and Robertson, G.L. (1994) Hypodipsic hypernatremia in a dog with defective osmoregulation of antidiuretic hormone. Journal of American Veterinary Medical Association 204, 922–925.
Dickinson, P.J., Sturges, B.K., Kass, P.H. and LeCouteur, R.A. (2006) Characteristics of cisternal cerebrospinal fluid associated with intracranial meningiomas in dogs: 56 cases (1985–2004). Journal of American Veterinary Medical Association 228, 564–567.
Diniz, P.P. and Breitschwerdt, E.B. (2012) Anaplasma phagocytophilum (Canine granulocytotropic anaplasmosis). In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 244–254.
Dow, S.W., Fettman, M.J., LeCouteur, R.A. and Allen, T.A. (1987) Hypodipsic hypernatremia and associated myopathy in a hydrocephalic cat with transient hypopituitarism. Journal of American Veterinary Medical Association 191, 217–221.
Dubey, J.P. and Lappin, M.R. (2012) Toxoplasmosis and neosporosis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 806–827.
Dugan, S.J., Schwarz, P.D., Roberts, S.M. and Ching, S.V. (1993) Primary optic nerve meningioma and pulmonary metastasis in a dog. Journal of the America Animal Hospital Association 29, 11–16.
Dunning, M.D., Lowrie, C.S., Bexfield, N.H., Dobson, J.M. and Herrtage, M.E. (2009) Exogenous insulin treatment after hypofractionated radiotherapy in cats with diabetes mellitus and acromegaly. Journal of Veterinary Internal Medicine 23, 243–249.
Elia, G., Decaro, N., Martella, V., Cirone, F., Lucente, M.S., Lorusso, E., Di Trani, L. and Buonavoglia, C. (2006) Detection of canine distemper virus in dogs by real-time RT-PCR. Journal of Virologic Methods 136, 171–176.
Elliott, D.A., Feldman, E.C., Koblik, P.D., Samii, V.F. and Nelson, R.W. (2000) Prevalence of pituitary tumors among diabetic cats with insulin resistance. Journal of American Veterinary Medical Association 216, 1765–1768.
Espino, L., Suarez, M., Santamarina, G., Vila, M., Miño, N. and Lopez-Peña, M. (2009) First report of the simultaneous occurrence of choroid plexus papilloma and meningioma in a dog. Acta Veterianaria Hungarica 57(3), 389–397.
Esteve-Ratsch, B., Kneissl, S. and Gabler, C. (2001) Comparative evaluation of the ventricles in the Yorkshire terrier and the German shepherd dog using low-field MRI. Journal of Veterinary Radiology and Ultrasound 42, 410–413.
Farias, F.H., Zeng, R., Johnson, G.S., Shelton, G.D., Paquette, D. and O’Brien, D.P. (2012) A L2HGDH initiator methionine codon mutation in a Yorkshire terrier with L-2-hydroxyglutaric aciduria. BMC Veterinary Research 26(8), 124.
Feliu-Pascual, A.L., Matiasek, K. and de Stefani, A. (2008) Efficacy of mycophenolate mofetil for the treatment of presumptive granulomatous meningoencephalomyelitis: preliminary results. Journal of Veterinary Internal Medicine 22, 509.
Ferguson, P.L., Smith, G.M. and Wannamaker, B.B. (2010) A population based study of risk of epilepsy after hospitalization for traumatic brain injury. Epilepsia 51, 891–898.
Flegel, T., Bottcher, I. and Matiasek, K. (2007) Treatment of immune-mediated non-infectious encephalitis: alternative lomustine. In: 20th Annual Symposium of the European College of Veterinary Neurology. Bern (Switzerland), 508 pp.
Foley, J.E., Lapointe, J.M., Koblik, P., Poland, A. and Pedersen, N.C. (1998) Diagnostic features of clinical neurologic feline infectious peritonitis. Journal of Veterinary Internal Medicine 12(6), 415–423.
Forterre, F., Fritsch, G., Kaiser, S., Matiasek, K. and Brunnberg, L. (2006) Surgical approach for tentorial meningiomas in cats: A review of six cases. Journal of Feline Medicine and Surgery 8, 227–233.
Forterre, F., Jaggy, A., Rohrbach, H., Dickomeit, M. and Konar, M. (2009) Modified temporal approach for a rostro-temporal basal meningioma in a cat. Journal of Feline Medicine and Surgery 11, 510–513.
Foster, E.S., Carrillo, J.M. and Patnaik, A. (1988) Clinical signs of tumours affecting the rostral cerebrum in 43 dogs. Journal of Veterinary Internal Medicine 2, 71–74.
Freeman, C. and Platt, S. (2012) Head trauma. In: Small Animal Neurological Emergencies. Manson Publishing, London, pp. 363–382.
Frey, L.C. (2003) Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia 44(Suppl. 10), 11–17.
Friedenberg, S.G., Butler, A.L., Wei, L., Moore, S.A. and Cooper, E.S. (2012) Seizures following head trauma in dogs: 259 cases (1999-2009). Journal of American Veterinary Medical Association 241(11), 1479–1483.
Friedman, A. and Dingledine, R. (2011) Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia 52(Suppl. 3), 33–39.
Fulton, L.M. and Steinberg, H.S. (1990) Preliminary study of lomustine in the treatment of intracranial masses in dogs following localization by imaging techniques. Seminars in Veterinary Medicine and Surgery (Small Animals) 5, 241–245.
Gallagher, J.G., Berg, J., Knowles, K.E., Williams, L.L. and Bronson, R.T. (1993) Prognosis after surgical excision of cerebral meningiomas in cats: 17 cases (1986–1992). Journal of the American Veterinary Medical Association 203, 1437–1440.
Gallagher, J.G., Penninck, D., Boudrieau, R.J., Schelling, S.H. and Berg, J. (1995) Ultrasonography of the brain and vertebral canal in dogs and cats: 15 cases (1988-1993). Journal of American Veterinary Medical Association 207, 1320–1324.
Garcia, R.S., Wheat, L.J., Cook, A.K., Kirsch, E.J. and Sykes, J.E. (2012) Sensitivity and specificity of a blood and urine galactomannan antigen assay for diagnosis of systemic aspergillosis in dogs. Journal of Veterinary Internal Medicine 26(4), 911–919.
Garosi, L. (2010) Cerebrovascular Disease in Dogs and Cats. The Veterinary Clinics of North America Small Animal Practice 40, 65–79.
Garosi, L., McConnell, J.E., Platt, S.R., Barone, G., Baron, J.C., de Lahunta, A. and Schatzberg, S.J. (2005a) Results of investigations and outcome of dog brain infarcts. Journal of Veterinary Internal Medicine 19, 725–731.
Garosi, L., Penderis, J., McConnell, J.F. and Jakobs, C. (2005b) L-2-hydroxyglutaric aciduria in a West Highland white terrier. The Veterinary Record 29, 156(5), 145–147.
Garosi, L., Dawson, A., Couturier, J., Matiasek, L., de Stefani, A., Davies, E., Jeffery, N. and Smith, P. (2010) Necrotizing cerebellitis and cerebellar atrophy caused by Neospora caninum infection: magnetic resonance imaging and clinicopathologic findings in seven dogs. Journal of Veterinary Internal Medicine 24(3), 571–578.
Geib, L.W. (1966) Ossifying meningioma with extracranial metastasis in a dog. Veterinary Pathology 3, 247–253.
Glass, E.N., Cornetta, A.M., de Lahunta, A., Center, S.A. and Kent, M. (1998) Clinical and clinicopathological features in 11 cats with Cuterebra larvae myiasis of the central nervous system. Journal of Veterinary Internal Medicine 12, 365–368.
Glass, E.N., Kapatkin, A., Vite, C. and Steinberg, S.A. (2000) A modified bilateral transfrontal sinus approach to the canine frontal lobe and olfactory bulb: Surgical technique and five cases. Journal of the American Animal Hospital Association 36, 43–50.
Gnirs, K. (2006) Ciclosporin treatment of suspected granulomatous meningoencephalomyelitis in three dogs. Journal of Small Animal Practice 47(4), 201–206.
Gonçalves, R., Carrera, I., Garosi, L., Smith, P.M., McConnell, F.J. and Penderis, J. (2011) Clinical and topographic magnetic resonance imaging characteristics of suspected thalamic infarcts in 16 dogs. Veterinary Journal 188(1), 39–43.
Gordon, L.E., Thacher, C., Matthiesen, D.T. and Joseph, R.J. (1994) Results of craniotomy for the treatment of cerebral meningioma in 42 cats. Veterinary Surgery 23, 94–100.
Graham, N.S., Crichton, S., Koutroumanidis, M., Wolfe, C.D. and Rudd, A.G. (2013) Incidence and associations of poststroke epilepsy: the prospective South London stroke register. Stroke 44(3), 605–611.
Structural Epilepsy
Granger, N., Smith, P.M. and Jeffery, N.D. (2010) Clinical findings and treatment of non-infectious meningoencephalomyelitis in dogs: A systematic review of 457 published cases from 1962 to 2008. The Veterinary Journal 184, 290–297.
Greco, J.J., Aiken, S.A., Berg, J.M., Monette, S. and Bergman, P.J. (2006) Evaluation of intracranial meningioma resection with a surgical aspirator in dogs: 17 cases (1996–2004). Journal of American Veterinary Medical Association 229, 394–400.
Gredal, H., Skerritt, G.C., Gideon, P., Arlien-Soeborg, P. and Berendt, M. (2013) Spontaneous ischaemic stroke in dogs: clinical topographic similarities to humans. Acta Neurologica Scandinavica doi: 10.1111/ ane.12092. [Epub ahead of print]
Greene, C.E. (2012a) Rabies and other Lyssavirus infections In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 179–197.
Greene, C.E. (2012b) Antifungal chemotherapy. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 579–586.
Greene, C.E. and Calpin, J. (2012) Antimicrobial drug formulary. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 1207–1320.
Greene, C.E. and Vandevelde, M. (2012) Canine distemper. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 25–42.
Greene, C.E., Kidd, L. and Breitschwerdt, E.B. (2012) Rocky Mountain and Mediterranean spotted fevers, cat-flea typhuslike disease, Ricketsial pox, and Typhus. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 259–270.
Greene, R.T. (2012) Coccidioidomycosis and paracoccidioidomycosis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 634–645.
Gregory, C.R., Stewart, A., Sturges, B., DeManvelle, T., Cannon, A., Ortega, T., Harb, M. and Morris, R.E. (1998) Leflunomide effectively treats naturally occurring immune-mediated and inflammatory diseases of dogs that are unresponsive to conventional therapy. Transplantion Proceedings 30, 4143–4148.
Griffin, J.F., Young, B.D. and Levine, J.M. (2009) Imaging diagnosis-chronic canine distemper meningoencephalitis. Veterinary Radiology and Ultrasound 50(2), 182–184.
Grohmann, K.S., Schmidt, M.J., Moritz, A. and Kramer, M. (2012) Prevalence of seizures in cats after head trauma. Journal of American Veterinary Medical Association 241(11), 1467–1470.
Gunn-Moore, D. and Reed, N. (2011) CNS disease in the cat. Current knowledge of infectious causes. Journal of Feline Medicine and Surgery 13, 824–836.
Haan, C.E., Kraft, S.L., Gavin, P.R., Wendling, L.R. and Griebenow, M.L. (1994) Normal variation in size of the lateral ventricles of the Labrador retriever dog as assessed by magnetic resonance imaging. Journal of Veterinary Radiology and Ultrasound 35, 83–86.
Hanson, J.M., Teske, E., Voorhout, G., Galac, S., Kooistra, H.S. and Meij, B.P. (2007) Prognostic factors for outcome after transsphenoidal hypophysectomy in dogs with pituitary-dependent hyperadrenocorticism. Journal of Neurosurgery 107(4), 830–840.
Harkin, K.R., Goggin, J.M. and DeBey, B.M. (1999) Magnetic resonance imaging of the brain of a dog with hereditary polioencephalomyelopathy. Journal of American Veterinary Medical Association 214(9), 1342–1344.
Harrus, S., Waner, T. and Neer, T.M. (2012) Ehrlichia canis infection. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 227–238.
Hartmann, K. (2005) Feline infectious peritonitis. The Veterinary Clinics of North America Small Animal Practice 35, 39–81.
Hayes, G.M. (2009) Severe seizures associated with traumatic brain injury managed by controlled hypothermia, pharmacologic coma, and mechanical ventilation in a dog. Journal of Veterinary Emergency and Critical Care 19(6), 629–634.
Heading, K.L., Brockley, L.K. and Bennett, P.F. (2011) CCNU (lomustine) toxicity in dogs: a retrospective study (2002-07). Australian Veterinary Journal 89(4), 109–116.
Headley, S.A., Scorpio, D.G., Vidotto, O. and Dumler, J.S. (2011) Neorickettsia helminthoeca and salmon poisoning disease: a review. The Veterinary Journal 187(2), 165–173.
Hecht, S. and Adams, W.H. (2010a) MRI of Brain Disease in Veterinary Patients Part 1: Basic Principles and Congenital Brain Disorders. The Veterinary Clinics of North America Small Animal Practice 40, 21–38.
Hecht, S. and Adams, W.H. (2010b) MRI of Brain Disease in Veterinary Patients Part 2: Acquired Brain Disorders. The Veterinary Clinics of North America Small Animal Practice 40, 39–63.
Hecht, S., Adams, W.H., Smith, J.R. and Thomas, W.B. (2011) Clinical and imaging findings in five dogs with intracranial blastomycosis (Blastomyces dermatiditis). Journal of American Animal Hospital Association 47(4), 241–249.
Heidner, G.L., Kornegay, J.N. and Page, R.L. (1991) Analysis of survival in a retrospective study of 86 dogs with brain tumors. Journal of Veterinary Internal Medicine 5, 219–226.
Henke, D. and Vandevelde, M. (2012) Pseudorabies. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier London, pp. 198–201.
Higginbotham, M.J., Kent, M. and Glass, E.N. (2007) Noninfectious inflammatory central nervous system diseases in dogs. Compendium on Continuing Education for the Practicing Veterinarian 29(8), 488–497.
Huifang, L., McDonald, W., Parada, I., Faria, L., Graber, K., Takahashi, K., Yunyong, M. and Prince, B. (2011) Targets for preventing epilepsy following cortical injury. Neuroscience Letters 27, 497(3), 172–176.
Hung, C. and Chen, J.W. (2012) Treatment of post-traumatic epilepsy. Current Treatment Options in Neurology 14(4), 293–306.
Ilha, M.R.S., Rajeev, S., Watson, C. and Woldemeskel, M. (2010) Meningoencephalitis caused by Mycoplasma edwardii in a dog. Journal of Veterinary Diagnostic Investigation 22, 805–808.
Jaderlund, K.H., Hansson, K., Berg, A.L., Sjöström, A. and Narfström, K. (2003) Cerebral ventricular size in developing normal kittens measured by ultrasonography. Journal of Veterinary Radiology and Ultrasound 44, 581–588.
Jaggy, A. and Vandevelde, M. (1988) Multisystem neuronal degeneration in cocker spaniels. Journal of Veterinary Internal Medicine 2, 117–120.
James, F.M. and Poma, R. (2010) Neurological manifestations of feline cuterebriasis. Canadian Veterinary Journal 51(2), 213–215.
Jull, P., Browne, E., Boufana, B.S., Schöniger, S. and Davies, E. (2012) Cerebral coenurosis in a cat caused by Taenia serialis: neurological, magnetic resonance imaging and pathological features. Journal of Feline Medicine and Surgery 2012 Sep;14(9):646-9. doi: 10.1177/1098612X12458211.
Jung, D.I., Kim, H.J., Park, C., Kim, J.W., Kang, B.T., Lim, C.Y., Park, E.H., Sur, J.H., Seo, M.H., Hahm, D.H. and Park, H.M. (2006) Long-term chemotherapy with lomustine of intracranial meningioma occurring in a miniature schnauzer. The Journal of Veterinary Medical Science 68, 383–386.
Jungehulsing, G.J., Heuschmann, P.U., Holtkamp, M., Schwab, S. and Kolominsky-Rabas, P.L. (2013) Incidence and predictors of post-stroke epilepsy. Acta Neurologica Scandinavica doi: 10.1111/ane.12070.
Kaser-Hotz, B., Rohrer, C.R. and Stankeova, S. (2002) Radiotherapy of pituitary tumours in five cats. Journal of Small Animal Practice 43, 303–307.
Kelmer, E., Shelton, G.D., Williams, D.A., Ruaux, C.G., Kerl, M.E. and O’Brien, D.P. (2007) Organic acidemia in a young cat associated with cobalamin deficiency. Journal of Veterinary Emergency and Critical Care 17, 299–304.
Kent, M. (2009) The cat with neurological manifestations of systemic disease: key conditions impacting on the CNS. Journal of Feline Medicine and Surgery 11, 395–407.
Kent, M. (2012) Bacterial infections in the central nervous system. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 1045–1058.
Kent, M.S., Bommarito, D., Feldman, E. and Theon, A.P. (2007) Survival, neurologic response, and prognostic factors in dogs with pituitary masses treated with radiation therapy and untreated dogs. Journal of Veterinary Internal Medicine 21, 1027–1033.
Kerrigan, S. and Grant, R. (2011) Antiepileptic drugs for treating seizures in adults with brain tumours. Cochrane Database of Systematic Reviews 10(8).
Kii, S., Uzuka, Y., Taura, Y., Nakaichi, M., Takeuchi, A., Inokuma, H. and Onishi, T. (1997) Magnetic resonance imaging of the lateral ventricles in beagle-type dogs. Journal of Veterinary Radiology and Ultrasound 38, 430–433.
Klopp, L.S. and Rao, S. (2009) Endoscopic-assisted intracranial tumor removal in dogs and cats: long-term outcome of 39 cases. Journal of Veterinary Internal Medicine 23, 108–115.
Koch, M.W., Sánchez, M.D. and Long, S. (2011) Multifocal oligodendroglioma in three dogs. Journal American Animal Hospital Association 47(5), 77–85.
Kolata, R.J., Kraut, N.H. and Johnston, D.E. (1974) Patterns of trauma in urban dogs and cats: a study of 1,000 cases. Journal of American Veterinary Medical Association 164, 499–502.
Kostolich, M. and Dulisch, M.L. (1987) A surgical approach to the canine olfactory bulb for meningioma removal. Veterinary Surgery 16, 273–277.
Kramer, K., Schaudien, D., Eisel, U.L., Herzog, S., Richt, J.A., Baumgärtner, W. and Herden, C. (2012) TNFoverexpression in Borna disease virus-infected mouse brains triggers inflammatory reaction and epileptic seizures. PLoS One 7(7): e41476.
Structural Epilepsy
Kube, S.A., Bruyette, D.S. and Hanson, S.M. (2003) Astrocytomas in young dogs. Journal American Animal Hospital Association 39(3), 288–293.
Lackay, S.N., Kuang, Y. and Fu, Z.F. (2008) Rabies in small animals. The Veterinary Clinics of North America Small Animal Practice 38(4), 851–861.
Lane, L.V., Meinkoth, J.H., Brunker, J., Smith, S.K., Snider, T.A., Thomas, J., Bradway, D. and Love, B.C. (2012) Disseminated protothecosis diagnosed by evaluation of CSF in a dog. Veterinary Clinical Pathology 41(1), 147–152.
Legendre, A.M. (2012) Blastomycosis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 606–614.
Lester, N.V., Hopkins, A.L., Bova, F.J., Friedman, W.A., Buatti, J.M., Meeks, S.L. and Chrisman, C.L. (2001) Radiosurgery using a stereotactic headframe system for irradiation of brain tumors in dogs. Journal of American Veterinary Medical Association 219(1550), 1562–1567.
Lipitz, L., Rylander, H., Forrest, L.J. and Foy, D.S. (2010) Clinical and magnetic resonance imaging features of central nervous system blastomycosis in 4 dogs. Journal of Veterinary Internal Medicine 24(6), 1509–1514.
Lorenz, M.D., Coates, J. and Kent, M. (2011) Handbook of Veterinary Neurology, 5th edn. Saunders-Elsevier, pp. 451–475.
Louis, D.N., Ohgaki, H., Wiestler, O.D., Cavenee, W.K., Burger, P.C., Jouvet, A., Scheithauer, B.W. and Kleihues, P. (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathologica 114, 97–109.
Lowenstein, D.H. (2009) Epilepsy after head injury: an overview. Epilepsia 50(Suppl. 2), 4–9.
Lowrie, M., De Risio, L., Dennis, R., Llabrés-Díaz, F. and Garosi, L. (2012) Concurrent medical conditions and long-term outcome in dogs with non-traumatic intracranial hemorrhage. Veterinary Radiology and Ultrasound 53(4), 381–388.
Lowrie, M., Smith, P.M. and Garosi, L. (2013) Meningoencephalitis of unknown origin: investigation of prognostic factors and outcome using a standard treatment protocol. The Veterinary Record 5. [Epub ahead of print]
MacKillop, E. (2011) Magnetic resonance imaging of intracranial malformations in dogs and cats. Journal of Veterinary Radiology and Ultrasound 52(1)(Suppl. 1), S42–51.
Márquez, M., Ródenas, S., Molin, J., Rabanal, R.M., Fondevila, D., Añor, S. and Pumarola, M. (2012) Protothecal pyogranulomatous meningoencephalitis in a dog without evidence of disseminated infection. The Veterinary Record 171(4), 100.
Martella, V., Elia, G. and Buonavoglia, C. (2008) Canine Distemper Virus. The Veterinary Clinics of North America Small Animal Practice 38, 787–797.
Martin-Vaquero, P., da Costa, R.C., Wolk, K.E., Premanandan, C. and Oglesbee, M.J. (2012) MRI features of gliomatosis cerebri in a dog. Veterinary Radiology and Ultrasound 53(2), 189–192.
Maschio, M. (2012) Brain tumor-related epilepsy. Current Neuropharmacology 10(2), 124–133.
McDonnell, J.J., Kalbko, K., Keating, J.H., Sato, A.F. and Faissler, D. (2007) Multiple meningiomas in three dogs. Journal of the American Animal Hospital Association 43, 201–208.
McDonough, S.P., Van Winkle, T.J., Valentine, B.A., vanGessel, Y.A. and Summers, B.A. (2002) Clinicopathological and immunophenotypical features of canine intravascular lymphoma (malignant angioendotheliomatosis). Journal of Comparative Pathology 126, 277–288.
Meij, B., Voorhout, G. and Rijnberk, A. (2002) Progress in transsphenoidal hypophysectomy for treatment of pituitary-dependent hyperadrenocorticism in dogs and cats. Molecular and Cellular Endocrinology 197, 89–96.
Menaut, P., Landart, J., Behr, S., Lanore, D. and Trumel, C. (2008) Treatment of 11 dogs with meningoencephalomyelitis of unknown origin with a combination of prednisolone and cytosine arabinoside. The Veterinary Record 162, 241–245.
Menon, B. and Shorvon, S.D. (2009) Ischaemic stroke in adults and epilepsy. Epilepsy Research 87(1), 1–11.
Messer, J.S., Wagner, S.O., Baumwart, R.D. and Colitz, C.M. (2008) A case of canine streptococcal meningoencephalitis diagnosed using universal bacterial polymerase chain reaction assay. Journal American Animal Hospital Association 44(4), 205–209.
Michael, B.D. and Solomon, T. (2012) Seizures and encephalitis: clinical features, management, and potential pathophysiologic mechanisms. Epilepsia 53(Suppl. 4), 63–71.
Morrison, J.P., Schatzberg, S.J., de Lahunta, A., Ross, J.T., Bookbinder, P. and Summers, B.A. (2006) Oligodendroglial dysplasia in two bullmastiff dogs. Veterinary Pathology 43(1), 29–35.
Motta, L., Mandara, M.T. and Skerritt, G.C. (2012) Canine and feline intracranial meningiomas: An updated review. The Veterinary Journal 192(2), 153–165.
Munana, K.R. and Luttgen, P.J. (1998) Prognostic factors for dogs with granulomatous meningoencephalomyelitis: 42 cases (1982–1996). Journal of American Veterinary Medical Association 212, 1902–1906.
Negrin, A., Lamb, C.R., Cappello, R. and Cherubini, G.B. (2007) Results of magnetic resonance imaging in 14 cats with meningoencephalitis. Journal of Feline Medicine and Surgery 9(2), 109–116.
Nghiem, P.P. and Schatzberg, S.J. (2010) Conventional and molecular diagnostic testing for the acute neurologic patient. Journal of Veterinary Emergency and Critical Care 20(1), 46–61.
Noonan, M., Kline, K.L. and Meleo, K. (1997) Lymphoma of the central nervous system: a retrospective study of 18 cats. Compendium on Continuing Education for the Practicing Veterinarian 19, 497–504.
O’Brien, D.P., Barshop, B.A., Faunt, K.K., Johnson, G.C., Gibson, K.M. and Shelton, G.D. (1999) Malonic aciduria in Maltese dogs: normal methylmalonic acid concentrations and malonyl-CoA decarboxylase activity in fibroblasts. Journal of Inherited Metabolism Disease 22, 883–890.
O’Brien, S.J., Troyer, J.L., Brown, M.A., Johnson, W.E., Antunes, A., Roelke, M.E. and Pecon-Slattery, J. (2012) Emerging viruses in the Felidae: shifting paradigms. Viruses 4(2), 236–257.
Packer, R.A., Cohn, L.A., Wohlstadter, D.R., Shelton, G.D., Naylor, J.M., Zello, G.A., Ewaschuk, J.B., Williams, D.A., Ruaux, C.G. and O’Brien, D.P. (2005) D-lactic acidosis secondary to exocrine pancreatic insufficiency in a cat. Journal of Veterinary Internal Medicine 19, 106–110.
Pákozdy, A., Leschnik, M., Tichy, A.G. and Thalhammer, J.G. (2008) Retrospective clinical comparison of idiopathic versus symptomatic epilepsy in 240 dogs with seizures. Acta Veterinaria Hungarica 56(4), 471–483.
Pákozdy, A., Leschnik, M., Kneissl, S., Gumpenberger, M., Gruber, A., Tichy, A. and Thalhammer, J.G. (2009) Improved survival time in dogs with suspected GME treated with ciclosporin. Veterinary Record 17, 164(3), 89–90.
Palus, V., Volk, H.A., Lamb, C.R., Targett, M.P. and Cherubini, G.B. (2011) MRI Features of CNS lymphoma in dogs and cats. Veterinary Radiology and Ultrasound 8, 1–6.
Paul, A.E., Lenard, Z. and Mansfield D.C.S. (2010) Computed tomography diagnosis of eight dogs with brain infarction. Australian Veterinary Journal 88, 374–380.
Pedersen, N.C. (2009) A review of feline infectious peritonitis virus infection: 1963–2008. Journal of Feline Medicine and Surgery 11, 225–258.
Penderis, J., Calvin, J., Abramson, C., Jakobs, C., Pettitt, L., Binns, M.M., Verhoeven, N.M., O’Driscoll, E., Platt, S.R. and Mellersh, C.S. (2007) L-2-hydroxyglutaric aciduria: characterisation of the molecular defect in a spontaneous canine model. Journal of Medical Genetics 44, 334–340.
Pérez, V., Vidal, E., González, N., Benavides, J., Ferreras, M.C., Villagrasa, M. and Pumarola, M. (2005) Orbital meningioma with a granular cell component in a dog, with extracranial metastasis. Journal of Comparative Pathology 133, 212–217.
Pesteanu-Somogyi, L.D., Radzai, C. and Pressler, B.M. (2006) Prevalence of feline infectious peritonitis in specific cat breeds. Journal of Feline Medicine and Surgery 8, 1–5.
Platt, S.R. and Haag, M. (2002) Canine status epilepticus: A retrospective study of 50 cases. Journal of Small Animal Practice 43, 151–153.
Platt, S.R., Radaelli, S.T. and McDonnell, J.J. (2001) The prognostic value of the modified Glasgow coma scale in head trauma in dogs. Journal of Veterinary Internal Medicine 15, 581–584.
Platt, S.R., Scase, T.J., Adams, V., Wieczorek, L., Miller, J., Adamo, F. and Long, S. (2006) Vascular endothelial growth factor expression in canine intracranial meningiomas and association with patient survival. Journal of Veterinary Internal Medicine 20, 663–668.
Platt, S., Adams, V., McConnell, F., Matiasek, L., de Stefani, A., Pascual, A. and De Risio, L. (2007) Magnetic resonance imaging imaging evaluation of head trauma in 32 dogs; associations with modified Glasgow coma score and patient outcome. Journal of Veterinary Internal Medicine 21, 1145.
Polizopoulou, Z.S., Koutinas, A.F., Souftas, V.D., Kaldrymidou, E., Kazakos, G. and Papadopoulos, G. (2004) Diagnostics correlation of CT-MRI and histopathology in 10 dogs with brain neoplasms. Journal of Veterinary Medicine, A, Physiology, Pathology, Clinical Medicine 51(5), 226–231.
Pressler, B.M. (2012) Protothecosis and Chlorellosis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 696–701.
Prince, D.A., Parada, I., Scalise, K., Graber, K., Jin, X. and Shen, F. (2009) Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia 50(Suppl. 2), 30–40.
Quesnel, A.D., Parent, J.M., McDonell, W., Percy, D. and Lumsden, J.H. (1997) Diagnostic evaluation of cats with seizure disorders: 30 cases (1991–1993). Journal of the American Veterinary Medical Association 210, 65–71.
Structural Epilepsy
Radaelli, S.T. and Platt, S.R. (2002) Bacterial Meningoencephalomyelitis in Dogs: A Retrospective Study of 23 Cases (1990–1999). Journal of Veterinary Internal Medicine 16, 159–163.
Riviere, J.E. and Papich, M.G. (2009) Chemotherapy of microbial diseases. In: Veterinary Pharmacology and Therapeutics, 9th edn. Wiley Blackwell, pp. 817–1012.
Robson, K. and Smith, P.M. (2011) Cryptococcal meningoencephalitis in a dog. Veterinary Record 21, 168(20), 538.
Ródenas, S., Pumarola, M., Gaitero, L., Zamora, A. and Añor, S. (2011) Magnetic resonance imaging findings in 40 dogs with histologically confirmed intracranial tumours. The Veterinary Journal 187, 85–91.
Rossmeisl, J.H., Jones, J.C., Zimmerman, K.L. and Robertson, J.L. (2013) Survival time following hospital discharge in dogs with palliatively treated primary brain tumors. Journal of American Veterinary Medical Association 15, 242(2), 193–198.
Rudà, R., Trevisan, E. and Soffietti, R. (2010) Epilepsy and brain tumors. Current Opinion in Oncology 22, 611–620.
Rudmann, D.G., Kazacos, K.R., Storandt, S.T., Harris, D.L. and Janovitz, E.B. (1996) Baylisascaris procyonis larva migrans in a puppy: a case report and update for the veterinarian. Journal of the American Animal Hospital Association 32(1), 73–76.
Rusbridge, C. and Wilkins, P. (2002) Neuronal heterotopia in a Lagotto Romagnola dog. In: Proceedings of ESVN, 15th Annual Symposium.
Saito, T.B., Alfieri, A.A., Wosiacki, S.R., Negrão, F.J., Morais, H.S. and Alfieri, A.F. (2006) Detection of canine distemper virus by reverse transcriptase-polymerase chain reaction in the urine of dogs with clinical signs of distemper encephalitis. Research in Veterinary Science 80, 116–119.
Salvadori, C., Gandini, G., Ballarini, A. and Cantile, C. (2008) Protothecal granulomatous meningoencephalitis in a dog. Journal of Small Animal Practice 49(10), 531–535.
Sanchez-Masian, D.F., Artuch, R., Mascort, J., Jakobs, C., Salomons, G., Zamora, A., Casado, M., Fernandez, M., Recio, A. and Lujan, A. (2012) L-2-hydroxyglutaric aciduria in two female Yorkshire terriers. Journal of American Animal Hospital Association 48(5), 366–371.
Sande, A. (2010) Traumatic brain injury: a review of pathophysiology and management. Journal of Veterinary Emergency and Critical Care 20(2), 177–190.
Schierhout, G. and Roberts, I. (2012) Withdrawn: Antiepileptic drugs for preventing seizures following acute traumatic brain injury. Cochrane Database of Systematic Reviews 13; 6, CD000173.
Schmidt, M.J., Klumpp, S., Amort, K., Jawinski, S. and Kramer, M. (2012) Porencephaly in dogs and cats: magnetic resonance imaging findings and clinical signs. Journal of Veterinary Radiology and Ultrasound 53(2), 142–149.
Schriefl, S., Steinberg, T.A., Matiasek, K., Ossig, A., Fenske, N. and Fischer, A. (2008) Etiologic classification of seizures, signalment, clinical signs, and outcome in cats with seizure disorders: 91 cases (2000–2004). Journal of the American Veterinary Medical Association 233, 1591–1597.
Schulman, F.Y., Ribas, J.L., Carpenter, J.L., Sisson, A.F. and LeCouteur, R.A. (1992) Intracranial meningioma with pulmonary metastasis in three dogs. Veterinary Pathology 29, 196–202.
Schwartz, M., Lamb, C.R., Brodbelt, D.C. and Volk, H.A. (2011) Canine intracranial neoplasia: clinical risk factors for development of epileptic seizures. Journal of Small Animal Practice 52(12), 632–637.
Schwartz-Porsche, D. and Kaiser, E. (1989) Feline epilepsy. Problems in Veterinary Medicine 1, 628–649.
Scott-Moncrieff, J.C., Chan, T.C., Samuels, M.L., Cook, J.R., Coppoc, G.L., DeNicola, D.B. and Richardson, R.C. (1991) Plasma and cerebrospinal fluid pharmacokinetics of cytosine arabinoside in dogs. Cancer Chemotherapy and Pharmacology 29, 13–18.
Selby, L.A., Hayes, H.M. and Becker, S.V. (1979) Epizootiologic features of canine hydrocephalus. American Journal of Veterinary Research 40(3), 411–413.
Sewell, A.C., Haskins, M.E. and Giger, U. (2007) Inherited metabolic disease in companion animals: searching for nature’s mistakes. The Veterinary Journal 174(2), 252–259.
Shihab, N., Davies, E., Kenny, P.J., Loderstedt, S. and Volk, H.A. (2011) Treatment of hydrocephalus with ventriculoperitoneal shunting in twelve dogs. Veterinary Surgery 40(4), 477–484.
Shores, A. (1989) Craniocerebral trauma. In: Current Veterinary Therapy X. W.B. Saunders, Philadelphia, pp. 847–854.
Simpson, K., Battersby, I. and Lowrie, M. (2012) Suspected acquired hypocobalaminaemic encephalopathy in a cat: resolution of encephalopathic signs and MRI lesions subsequent to cobalamin supplementation. Journal of Feline Medicine and Surgery 14(5), 350–355.
Skelly, B.J. and Franklin, R.J.M. (2002) Recognition and diagnosis of lysosomal storage diseases in the cat and dog. Journal of Veterinary Internal Medicine 16, 133–141.
Slapø, G.D., Lossius, M.I. and Gjerstad, L. (2006) Poststroke epilepsy: occurrence, predictors and treatment. Expert Review of Neurotherapeutics 6(12), 1801–1809.
Smith, M.O., Turrel, J.M., Bailey, C.S. and Gain, G.R. (1989) Neurologic abnormalities as the predominant signs of neoplasia of the nasal cavity in dogs and cats: seven cases (1973–1986). Journal of American Veterinary Medical Association 195, 242–245.
Smith, P.M., Haughland, S.P. and Jeffery, N.D. (2007) Brain abscess in a dog immunosuppressed using cyclosporin. The Veterinary Journal 173(3), 675–678.
Smith, P.M., Stalin, C.E., Shaw, D., Granger, N. and Jeffery, N.D. (2009) Comparison of two regimens for the treatment of meningoencephalomyelitis of unknown etiology. Journal of Veterinary Internal Medicine 23(3), 520–526.
Snyder, J.M., Shofer, F.S., Van Winkle, T.J. and Massicotte, C. (2006) Canine intracranial primary neoplasia: 173 cases (1986–2003). Journal of Veterinary Internal Medicine 20, 669–675.
Snyder, J.M., Lipitz, L., Skorupski, K.A., Shofer, F.S. and Van Winkle, T.J. (2008) Secondary intracranial neoplasia in the dog: 177 cases (1986–2003). Journal of Veterinary Internal Medicine 22, 172–177.
Spaulding, K.A. and Sharp, N.J.H. (1990) Ultrasonographic imaging of the lateral cerebral ventricles in the dog. Journal of Veterinary Radiology and Ultrasound 31, 59–64.
Spugnini, E.P., Thrall, D.E., Price, G.S., Sharp, N.J., Munana, K. and Page, R.L. (2000) Primary irradiation of canine intracranial masses. Veterinary Radiology and Ultrasound 41, 377–380.
Steinmetz, S., Tipold, A. and Löscher, W. (2013) Epilepsy after head injury in dogs: A natural model of post-traumatic epilepsy. Epilepsia doi: 10.1111/epi.12071.
Stoica, G., Levine, J., Wolff, J. and Murphy, K. (2011) Canine astrocytic tumors: a comparative review. Veterinary Pathology 48(1), 266–275.
Streeter, E.M., Rozanski, E.A. and Laforcade-Buress, A.D. (2009) Evaluation of vehicular trauma in dogs: 239 cases (January-December 2001). Journal of American Veterinary Medical Association 235, 405–408.
Sturges, B.K., Dickinson, P.J., Kortz, G.D., Berry, W.L., Vernau, K.M., Wisner, E.R. and LeCouteur, R.A. (2006) Clinical signs, magnetic resonance imaging features, and outcome after surgical and medical treatment of otogenic intracranial infection in 11 cats and 4 dogs. Journal of Veterinary Internal Medicine 20, 648–656.
Sturges, B.K., Dickinson, P.J., Bollen, A.W., Koblik, P.D., Kass, P.H., Kortz, G.D., Vernau, K.M., Knipe, M.F., Lecouteur, R.A. and Higgins, R.J. (2008) Magnetic resonance imaging and histological classification of intracranial meningiomas in 112 dogs. Journal of Veterinary Internal Medicine 22, 586–595.
Summers, B.A., Cummings, J.F. and DeLahunta, A. (2005) Tumors of the central nervous system. In: Veterinary Neuropathology. Mosby, St Louis.
Sykes, J.E. and Malik, R. (2012) Cryptococcosis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 621–634.
Sykes, J.E., Marks, S.L., Mapes, S., Schultz, R.M., Pollard, R.E., Tokarz, D., Pesavento, P.P., Lindsay, L.A. and Foley, J.E. (2010a) Salmon poisoning disease in dogs: 29 cases. Journal of Veterinary Internal Medicine 24(3), 504–513.
Sykes, J.E., Sturges, B.K., Cannon, M.S., Gericota, B., Higgins, R.J., Trivedi, S.R., Dickinson, P.J., Vernau, K.M., Meyer, W. and Wisner, E.R. (2010b) Clinical signs, imaging features, neuropathology, and outcome in cats and dogs with central nervous system cryptococcosis from California. Journal of Veterinary Internal Medicine 24(6), 1427–1438.
Syring, R.S., Otto, C.M. and Drobatz, K.J. (2001) Hyperglycemia in dogs and cats with head trauma: 122 cases (1997-1999). Journal of American Veterinary Medical Association 218(7), 1124–1129.
Talarico, L.R. and Schatzberg, S.J. (2010) Idiopathic granulomatous and necrotising inflammatory disorders of the canine central nervous system: a review and future perspectives. Journal of Small Animal Practice 51(3), 138–149.
Tamura, S., Tamura, Y., Ohoka, A., Hasegawa, T. and Uchida, K. (2007) A canine case of skull base meningioma treated with hydroxyurea. The Journal of Veterinary Medical Science 69, 1313–1315.
Théon, A.P. and Feldman, E.C. (1998) Megavoltage irradiation of pituitary macrotumors in dogs with neurologic signs. Journal of American Veterinary Medical Association 213, 225–231.
Théon, A.P., Lecouteur, R.A., Carr, E.A. and Griffey, S.M. (2000) Influence of tumor cell proliferation and sex-hormone receptors on effectiveness of radiation therapy for dogs with incompletely resected meningiomas. Journal of the American Veterinary Medicine Association 216, 701–707.
Thomas, W.B. (2010) Hydrocephalus in Dogs and Cats. The Veterinary Clinics of North America Small Animal Practice 40, 143–159.
Structural Epilepsy
Thomas, W.B., Adams, W.H., McGavin, M.D. and Gompf, R.E. (1997) Magnetic resonance imaging appearance of intracranial haemorrhage secondary to cerebral vascular malformation in a dog. Veterinary Radiology and Ultrasound 38, 371–375.
Timmann, D., Cizinauskas, S., Tomek, A., Doherr, M., Vandevelde, M. and Jaggy, A. (2008) Retrospective analysis of seizures associated with feline infectious peritonitis in cats. Journal of Feline Medicine and Surgery 10(1), 9–15.
Tipold, A. (2003) Cerebrospinal Fluid. In: Braund, K.G. (ed.) Clinical Neurology in Small Animals - Localization, Diagnosis and Treatment . International Veterinary Information Service (www.ivis.org), Ithaca, New York.
Tipold, A. and Stein, V.M. (2010) Inflammatory Diseases of the Spine in Small Animals. The Veterinary Clinics of North America Small Animal Practice 40, 871–879.
Tipold, A. and Vandevelde, M. (2012) Central European tick-borne encephalitis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 217–218.
Tipold, A., Vandevelde, M. and Jaggy, A. (1992) Neurological manifestations of canine distemper virus. Journal of Small Animal Practice 33, 466–470.
Tomek, A., Cizinauskas, S., Doherr, M., Gandini, G. and Jaggy, A. (2006) Intracranial neoplasia in 61 cats: localisation, tumour types and seizure patterns. Journal of Feline Medicine and Surgery 8(4), 243–253.
Triolo, A.J., Howard, M.O. and Miles, K.G. (1994) Oligodendroglioma in a 15-month-old dog. Journal of American Veterinary Medical Association 205(7), 986–988.
Troxel, M.T., Vite, C.H., Van Winkle, T.J., Newton, A.L., Tiches, D., Dayrell-Hart, B., Kapatkin, A.S., Shofer, F.S. and Steinberg, S.A. (2003) Feline intracranial neoplasia: retrospective review of 160 cases (1985–2001). Journal of Veterinary Internal Medicine 17, 850–859.
Troxel, M.T., Vite, C.H., Massicotte, C., McLear, R.C., Van Winkle, T.J., Glass, E.N., Tiches, D. and Dayrell-Hart, B. (2004) Magnetic resonance imaging features of feline intracranial neoplasia: retrospective analysis of 46 cats. Journal of Veterinary Internal Medicine 18(2), 176–189.
Turrel, J.M., Fike, J.R., LeCouteur, R.A., Pflugfelder, C.M. and Borcich, J.K. (1984) Radiotherapy of brain tumors in dogs. Journal of American Veterinary Medical Association 184, 82–86.
Uriarte, A., Moissonnier, P., Thibaud, J.L., Reyes-Gomez, E., Devauchelle, P. and Blot, S. (2011) Surgical treatment and radiation therapy of frontal lobe meningiomas in 7 dogs. The Canadian Veterinary Journal 52(7), 748–752.
Uriarte, J.L., Thibaud, K. and Gnirs, S. (2008) Lomustine treatment in noninfectious meningoencephalitis in 8 dogs. Journal of Veterinary Internal Medicine 22, 509.
Vaden, S.L., Wood, P.A., Ledley, F.D., Cornwell, P.E., Miller, R.T. and Page, R. (1992) Cobalamin deficiency associated with methylmalonic acidemia in a cat. Journal of American Veterinary Medical Association 200, 1101–1103.
Van de Beek, D. (2009) Corticosteroids for acute adult bacterial meningitis. Medecine et Maladies Infectiesues 39(7–8), 531–538.
Van de Beek, D., Brouwer, M.C., Thwaites, G.E. and Tunkel, A.R. (2012) Advances in treatment of bacterial meningitis. Lancet 380(9854), 1693–1702.
Van der Vlugt-Meijer, R.H., Meij, B.P. and Voorhout, G. (2007) Dynamic helical computed tomography of the pituitary gland in healthy dogs. Veterinary Radiology and Ultrasound 48, 118–124.
Vandevelde, M. (1984) Brain tumors in domestic animals: an overview. In: Proceedings of a Conference on Brain Tumors in Man and Animals, Research Triangle Park, North Carolina.
Vezzani, A., Aronica, E., Mazarati, A. and Pittman, Q.J. (2011) Epilepsy and brain inflammation. Experimental Neurology 1.
Victor, M. and Ropper, A.H. (2001) Cerebrovascular diseases. In: Adams and Victor’s Principles of Neurology. McGraw-Hill, New York, pp. 821–924.
Vinchon, M., Rekate, H. and Kulkarni, A.V. (2012) Pediatric hydrocephalus outcomes: a review. Fluids and Barriers of the CNS 27, 9(1), 18.
Vite, C.H. (2005) Neoplasia of the Nervous System In: Vite, C.H. (ed.) Braund’s Clinical Neurology in Small Animals: Localization, Diagnosis and Treatment . International Veterinary Information Service (www.ivis.org), Ithaca, New York.
Vite, C.H., Insko, E.K., Schotland, H.M., Panckeri, K. and Hendricks, J.C. (1997) Quantification of cerebral ventricular volume in English bulldogs. Journal of Veterinary Radiology and Ultrasound 38, 437–443.
Volk, A.V., Volk, H.A., Rest, J.R., Loderstedt, S. and Bond, R. (2012) Calcinosis cutis at cytarabine injection site in three dogs receiving prednisolone. The Veterinary Record 29, 171(13), 327.
Walmsley, G.L., Chandler, K., Davies, E.S., Lamb, C.R., Smyth, B. and Summers, B.A. (2009) Multi-focal cerebral oligoastrocytoma in a puppy. Journal of Small Animal Practice 50(8), 435–439.
Wensman, J.J., Berg, A.L. and Berg, B. (2012) Borna disease meningo encephalitis. In: Infectious Diseases of the Dog and Cat, 4th edn. Saunders-Elsevier, London, pp. 177–179.
Wessmann, A., Chandler, K. and Garosi, L. (2009) Ischaemic and haemorrhagic stroke in the dog. The Veterinary Journal 180, 290–303.
Westworth, D.R., Dickinson, P.J., Vernau, W., Johnson, E.G., Bollen, A.W., Kass, P.H., Sturges, B.K., Vernau, K.M., Lecouteur, R.A. and Higgins, R.J. (2008) Choroid plexus tumors in 56 dogs (1985–2007). Journal of Veterinary Internal Medicine 22, 1157–1165.
Williams, K.J., Summers, B.A. and de Lahunta, A. (1998) Cerebrospinal cuterebriasis in cats and its association with feline ischemic encephalopathy. Veterinary Pathology 35, 330–333.
Windsor, R.C., Sturges, B.K., Vernau, K.M. and Vernau, W. (2009) Cerebrospinal fluid eosinophilia in dogs. Journal of Veterinary Internal Medicine 23(2), 275–281.
Wisner, E.R., Dickinson, P.J. and Higgins, R.J. (2011) Magnetic resonance imaging features of canine intracranial neoplasia. Veterinary Radiology and Ultrasound 52(1)(Suppl. 1), 52–61.
Wolff, C.A., Holmes, S.P., Young, B.D., Chen, A.V., Kent, M., Platt, S.R., Savage, M.Y., Schatzberg, S.J., Fosgate, G.T. and Levine, J.M. (2012) Magnetic resonance imaging for the differentiation of neoplastic, inflammatory, and cerebrovascular brain disease in dogs. Journal of Veterinary Internal Medicine 26(3), 589–597.
Wong, M., Glass, E., de Lahunta, A. and Jackson, B. (2011) Intracranial anaplastic astrocytoma in a 19-weekold boxer dog. Journal of Small Animal Practice 52(6), 325–328.
You, G., Sha, Z. and Jiang, T. (2012) The pathogenesis of tumor-related epilepsy and its implications for clinical treatment. Seizure 21(3), 153–159.
Young, B.D., Levine, J.M., Porter, B.F., Chen-Allen, A.V., Rossmeisl, J.H., Platt, S.R., Kent, M., Fosgate, G.T. and Schatzberg, S.J. (2011) Magnetic resonance imaging features of intracranial astrocytomas and oligodendrogliomas in dogs. Veterinary Radiology and Ultrasound 52(2), 132–141.
Young, M., Bush, W., Sanchez, M., Gavin, P. and Williams, M. (2012) Serial MRI and CSF analysis in a dog treated with intrathecal amphotericin B for protothecosis. Journal of American Animal Hospital Association 48(2), 125–131.
Zaki, F.A. (1977) Spontaneous central nervous system tumors in the dog. The Veterinary Clinics of North America Small Animal Practice 7, 153–163.
Zaki, F.A. and Hurvitz, A.I. (1976) Spontaneous neoplasms of the central nervous system of the cat. Journal of Small Animal Practice 17, 773–782.
Zarfoss, M., Schatzberg, S., Venator, K., Cutter-Schatzberg, K., Cuddon, P., Pintar, J., Weinkle, T., Scarlett, J. and Delahunta, A. (2006) Combined cytosine arabinoside and prednisone therapy for meningoencephalitis of unknown aetiology in 10 dogs. Journal of Small Animal Practice 47, 588–595.
Zimmermann, R., Hülsmeyer, V., Sauter-Louis, C. and Fischer, A. (2009) Status epilepticus and epileptic seizures in dogs. Journal of Veterinary Internal Medicine 23(5), 970–976.
6 Idiopathic Epilepsy and Genetics
Simon Platt1 and Luisa De Risio2
Chronic, recurring seizure syndromes with no detectable underlying abnormalities are termed ‘idiopathic’ and are generally presumed to be genetic. The term idiopathic epilepsy is not applied simply to any patient in whom the cause of the seizures is unknown. Instead, it refers to recognized clinical syndromes with typical clinical features, such as age of onset and lack of other neurological abnormalities. Either of the newer classifications ‘genetic epilepsy’ or ‘unknown epilepsy’ could encompass idiopathic epilepsies (IEs). A diagnosis of IE can occur only in canine or feline patients after careful history-taking, physical and neurological examinations, blood chemistry tests, brain imaging and cerebrospinal fluid analysis have ruled out other causes of recurrent seizure activity. This is not dissimilar to human idiopathic (genetic) epilepsy where diagnosis is made based on electroencephalographic (EEG) examination, seizure type and age of onset (Asadi-Pooya et al., 2013). EEG-detected interictal paroxysmal activity has been documented in 25–86% of dogs with IE, consisting of single spikes, polyspikes and spike slow-wave complexes (see Chapter 11; Jaggy and Bernardini, 1998; Brauer et al., 2012).
Based on the strict definition of idiopathic epilepsy in which evidence of a pattern of inheritance must exist, idiopathic epilepsy in cats is rare (Quesnel et al., 1997; Barnes et al., 2004). However, in 21–59% of cats presenting for seizures, a cause of seizures is not identified and therefore may be referred to as idiopathic epilepsy (see Table 8.2).
Clinical Overview of Canine Idiopathic Epilepsy
The gross structure of the brain in animals with IE has no demonstrable pathologic lesions. The prevalence of epilepsy in dogs has been estimated at 1% to 5% (Holliday et al., 1970; Podell et al., 1995; Kearsley-Fleet et al., 2013). Although IE occurs in a number of species, the most comprehensive studies have been those of humans and dogs (Holliday et al., 1970; Podell et al., 1995; Berendt and Gram, 1999; Licht et al., 2002; Berendt et al., 2004). In order to establish a diagnosis, affected animals must have a normal neurologic examination and remain normal during the interictal period; however, recent work has questioned this dogma (see below). In addition, other systemic and brain abnormalities are not detected with diagnostic tests. Therefore from a clinical perspective, IE is a diagnosis of exclusion. The breed, age and history also provide important clues to an underlying hereditary basis, especially if a familial history of seizures exists.
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
Most dogs with IE suffer their first seizure between 6 months and 6 years of age, although seizures occasionally start before 6 months or as late as 10 years of age (Podell et al., 1995; Heynold et al., 1997; Jaggy and Bernardini, 1998; Berendt and Gram, 1999).
The finding of idiopathic seizures without an underlying cause does not necessarily rule in or out a genetic cause. Only careful breeding studies or pedigree analysis can prove a pattern of inheritance. The most common seizure type in dogs with IE is a focal seizure with secondary generalization (Berendt and Gram, 1999; Berendt et al., 2002, 2004, 2008; Licht et al., 2002, 2007). However, in some studies, generalized, tonic-clonic seizures were the most commonly observed seizure type (Heynold et al., 1997; Dichter, 2009). This may suggest the focal aspect of the seizure can be easily missed by the owner. IE seizure phenotypes vary among breeds; IE in some breeds manifests more frequently as focal onset seizures, whereas in others seizures are mostly generalized. The frequency of epileptic dogs suffering cluster seizures (CS) and status epilepticus (SE) also varies. A recent study of 407 IE dogs documented the frequency of CS to be 41% and the frequency of SE to be 2.5% (Monteiro et al., 2012). In this study, German shepherds and boxers were over-represented. Within specific breeds the prevalence of CS and SE can be very much higher. A recent study of Australian shepherds showed that 80% of the population suffered from SE or CS, very similar to the severe course of IE in other collie breeds such as Border collies (Weissl et al., 2012).
In the past, generalized tonic-clonic seizures were considered the most common type of seizure in dogs with IE, and it was even claimed focal-onset seizures were inconsistant with a diagnosis of IE. However, more recent observations reveal this is clearly not the case and dogs with IE can have a variety of focal-onset seizures, including secondarily generalized seizures, and some individuals have more than one type of seizure (Heynold et al., 1997; Jaggy and Bernardini, 1998; Patterson et al., 2003, 2005; Pákozdy et al., 2008; Gullov et al., 2011). The frequency of seizures varies tremendously, ranging from several a day to less than one a year (Podell et al., 1995; Heynold et al., 1997). Seizures are most common during rest or sleep (Pákozdy et al., 2008). Even though most seizures appear to occur spontaneously, they may be precipitated by a variety of factors. In human patients, sleep deprivation, emotional stress, menstruation, missed medication and concurrent illness are recognized (Haut et al., 2007). Similar factors are likely important in precipitating seizures in some animals. Reflex seizures are seizures that can be provoked by specific stimuli or events (Thomas, 2000). The most common trigger in people is flickering light, usually from a television. Other triggers include immersion in hot water, reading, certain sounds and eating. With reflex seizures, the trigger is specific and the latency between the trigger and seizure is short (seconds to minutes; Thomas, 2000). Infrequently an owner will describe a consistent temporal association between a specific sound and the onset of a seizure.
An inherited basis, familial transmission, or a higher incidence has been recognized in many breeds (Table 6.1). Any breed, including mix-breed dogs can be affected.
Interictal Signs
While the majority of canine IE patients are completely normal between seizures and do not display any other clinical signs, others may express mild abnormalities, such as episodic ataxia, between seizures (Jokinen et al., 2007). Likewise, human IE patients may also display such symptoms between seizures (Imbrici et al., 2004). In human medicine, an increasing number of studies have identified systemic and neurobehavioural or psychiatric illnesses associated with epilepsy (Gaitatzis et al., 2004; Tellez-Zenteno et al., 2005; Nuyen et al., 2006; Austin and Caplan, 2007; LaFrance et al., 2008). In fact in people with a history of major depression or anxiety there is an increased risk of unprovoked seizures and epilepsy confirming a bidirectional relationship (Heinrichs and Seyfried, 2006; Kanner, 2006). Neurobehavioural co-morbidities have also been reported and studied in a variety of rodent models of epilepsy (Heinrichs and Seyfried, 2006). In a recent study, neurobehavioural changes were found to be related not only to epilepsy but also to pharmacological response,
Table 6.1. Canine breeds in which a genetic component for idiopathic epilepsy has been proposed (Ekenstedt et al., 2012; Ekenstedt and Oberbauer, 2013).
Breed Reference
Australian shepherd Weissl et al. (2012) Beagle Bielfelt et al. (1971) Belgian shepherd Berendt et al. (2009);
Oberbauer et al. (2010);
Seppala et al. (2012) Belgian tervueren Oberbauer et al. (2003) Bernese mountain dog Kathmann et al. (1999) Border collie Hulsmeyer et al. (2010) Dalmatian Licht et al. (2002) English springer Patterson et al. (2005)
spaniel Finnish spitz Viitmaa et al. (2013) German shepherd dog Falco et al., (1974) Golden retriever Srenk and Jaggy (1996) Irish wolfhound Casal et al. (2006) Keeshond Hall and Wallace (1996) Labrador retriever Jaggy et al. (1999);
Berendt et al. (2002) Lagotto Romagnolo Jokinen et al. (2007) Petit Basset Griffon Gullov et al. (2011)
Vendeen Schipperke Koskinen et al. (2012) Shetland sheepdog Morita et al. (2002) Standard poodle Licht et al. (2007) Vizsla Patterson et al. (2003)
with pharmacoresistant rats having greater behaviour changes (Gastens et al., 2008).
A recent veterinary study investigated dogs with IE for associated behavioural changes (Shihab et al., 2011). The aim of the study was to look for behavioural changes associated with the development of epilepsy in dogs. Owners of a dog diagnosed with IE (n=80) completed a behavioural and seizure questionnaire. Drug-naïve dogs showed an increase in the behaviour factors: (i) Fear/Anxiety;
(ii) Defensive Aggression; and (iii) Abnormal Perception. In dogs receiving anti-epileptic medication (AEM), there were still increases in Fear/Anxiety and Abnormal Perception, but no longer in Defensive Aggression. Additionally increases in the following were observed:
(i)Abnormal Reactivity; (ii) Attachment Disorder;
(iii) Demented Behaviour; and (iv) Apathetic Behaviour. Pharmacoresistant dogs had larger increases in Controlling Aggression, Abnormal Perception and Demented Behaviour than those dogs, which were considered to be drug responders (Shihab et al., 2011).
Lifespan and Canine Idiopathic Epilepsy
Dogs suffering from IE have an increased risk of premature death compared with the general dog population (Proschowsky et al., 2003; Berendt et al., 2007; Hulsmeyer et al., 2010). This also applies for people with epilepsy for whom the risk of death is highest shortly after onset of seizures (Hauser et al., 1980; Neligan et al., 2011). The median survival time after the initial seizure is 2.07 years in Border collies and 2.3 years in a population of different purebred and mixed-breed dogs (Proschowsky et al., 2003; Berendt et al., 2007; Hulsmeyer et al., 2010). The life expectancy for Irish wolfhounds is shortened by almost 2 years in epileptic dogs compared with seizure-free relatives. The same study reported that more than 60% of the total number of deaths in the population were related to epilepsy (Casal et al., 2006). Factors such as gender, seizure onset, seizure frequency and seizure control might influence the lifespan of dogs with epilepsy. In one study, bitches lived longer with epilepsy compared with males, and daily treatment with anti-epileptic drugs did not influence the lifespan of dogs with epilepsy compared with dogs with epilepsy left untreated (Proschowsky et al., 2003). In Border collies, seizure onset before the age of 2 years has been shown to significantly decrease survival time (Hulsmeyer et al., 2010). A recent longitudinal study of survival in Belgian shepherds with IE revealed that the lifespan of epileptic dogs was not significantly shortened by the presence of epilepsy (Gullov et al., 2012). However, epilepsy was the predominant cause of death in the population (19/75 = 25%) and epilepsy-related deaths accounted for 70% (19/27) of all deaths in the group of dogs with epilepsy (Gullov et al., 2012). Two probable sudden unexpected deaths related to epilepsy occurred in dogs with generalized seizures. Sudden unexpected death in epilepsy (SUDEP) is the most common cause of death directly related to epilepsy in humans, with an increased frequency of unexpected deaths up to 24 times compared with unexpected deaths in non-epileptics (Ficker et al., 1998; Surges et al., 2009); The mechanisms of SUDEP remain elusive, and it seems unlikely that the identification of a single mechanism will explain all incidents, as SUDEP is more likely a result of several predisposing and triggering factors (Surges et al., 2009; Donner, 2011). Poorly controlled chronic epilepsy with tonic-clonic seizures seems to be the most well-defined risk factor for SUDEP in humans (Surges et al., 2009; Donner, 2011).
Genetics and Human Epilepsy
Epilepsies with complex inheritance
About 40% of human patients suffering from epilepsy have a genetic background that contributes to the aetiology of epilepsy (Gardiner, 2000). Most familial epilepsies like juvenile myoclonic epilepsy, childhood absence epilepsy and benign childhood epilepsy with centrotemporal spikes have a complex mode of inheritance resulting from the interaction of several loci together with environmental factors (McNamara, 1999). In patients with absence seizures (and their first degree relatives), biochemical changes (e.g. increased plasma glutamate levels) have been identified that can be related to a generalized increase in cortical excitability (van Gelder et al., 1980). Probably, the genetic predisposition of absence epilepsy is based on a gene-dependent biochemical derangement leading to increased cortical excitability. Genetic data generated by studies on animal models of absence epilepsy show a relative simple inheritance factor of one gene that determines being epileptic or not while other genes determine number and duration of epileptic seizures (Engelborghs et al., 2000).
Monogenic epilepsies
Monogenic epileptic disorders are rare, accounting for no more than 1% of patients. Recent advances in the genetics and molecular biology of these diseases unravelled the underlying pathophysiology of some of these epileptic syndromes. In 1996, Berkovic et al. described a new epileptic syndrome: familial temporal lobe epilepsy. Simple focal (partial) seizures with psychic or autonomic symptoms are frequently occurring seizure types whereas complex focal seizures are infrequent. Pedigree analysis suggested autosomal dominant inheritance with age-dependent penetrance (Berkovic et al., 1996). Linkage to chromosome 10q has been reported in one family but the genetic defect remains to be elucidated (Berkovic and Scheffer, 1997). Autosomal dominant focal epilepsy with auditory features is characterized by auditory hallucinations, although other sensory symptoms have been reported as well (Winawer et al., 2000). Clinical semiology points to a lateral temporal localization, which is supported by electroencephalogram (EEG) data that revealed inconsistant focal abnormalities over both temporal regions (Michelucci et al., 2000). In a single case, brain magnetic resonance imaging (MRI) showed atrophy with an increased T2 signal in the lateral portion of the right temporal lobe (Michelucci et al., 2000). This epileptic syndrome was found to be linked to chromosome 10q22–24 (Winawer et al., 2000). Gene defects underlying four other monogenic epilepsies (generalized epilepsy with febrile seizures, autosomal dominant nocturnal frontal lobe epilepsy, benign familial neonatal convulsions and episodic ataxia type I with focal seizures) have now been identified, shedding new light on the pathophysiology of epilepsy as these diseases are caused by ion channel mutations (Steinlein, 1998; Zuberi et al., 1999).
Generalized epilepsy with febrile seizures type I is an autosomal dominant epileptic syndrome that is caused by a point mutation in the b1-subunit of a voltage-gated Na+ channel (Wallace et al., 1998), whereas type II is caused by a point mutation in the a1-subunit of a voltage-gated Na+ channel (Escayg et al., 2000). These mutations cause distinct types of epilepsy in different members of the same family, which may result from inheritance of the mutant gene in the context of other susceptibility genes or environmental factors (McNamara, 1999).
Benign familial neonatal convulsions is a syndrome that is inherited in an autosomal dominant pattern. Mutations of two distinct but related voltage-gated K+ channel genes have been identified (Biervert et al., 1998). Although both genes (KCNQ2 and KCNQ3) are located on different chromosomes (20q and 8q respectively), their co-expression explains how these two different mutations cause an identical disease phenotype. In some families, autosomal dominant nocturnal frontal lobe epilepsy is caused by a point mutation in a gene on chromosome 20q (CHRNA4), encoding the a4 subunit of the neuronal nicotinic acetylcholine (ACh) receptor (Steinlein et al., 1995). At least some ACh receptors are located pre-synaptically, thus promoting the release of neurotransmitters such as GABA. The mutant receptor causes a reduction of ACh-mediated Ca2+ flux, which results in a decrease of GABA released from presynaptic terminals leading to synaptic disinhibition (McNamara, 1999). However, the majority of the families with autosomal dominant nocturnal frontal-lobe epilepsy are not linked to CHRNA4, indicating the presence of genetic heterogeneity (Gardiner, 2000).
Episodic ataxia type I is a rare autosomal dominant disorder, characterized by brief episodes of ataxia associated with myokymia (Zuberi et al., 1999). The patients suffering from this syndrome also show focal epileptic seizures. The syndrome is associated with point mutations in the human voltage-gated potassium channel gene on chromosome 12p13 (Zuberi et al., 1999). As potassium channels determine the excitability of neurons, it is suggested that this mutation is pathogenic (Zuberi et al., 1999). These recent discoveries illustrate that ion channel dysfunctions can play a crucial role in the pathophysiology of epilepsy. As several AEMs act on ion channels, these findings are relevant to other epileptic syndromes in man.
For a far more complete discussion of the genetics of human epilepsy, readers should consult recent reviews (Gardiner, 2005; Green-berg and Pal, 2007; Gurnett and Hedera, 2007; Baulac and Baulac, 2009). Despite these advances, the genetic bases for the majority of human IEs remain unsolved and are typically referred to as the common or complex non-Mendelian IE syndromes. The fact that replicate studies in different families/populations suffering similar types of seizures do not always reveal similar mutations reemphasizes that there are most likely many genes responsible for epilepsy.
Genetics and Canine Epilepsy
Dog breeds may be more likely to have a founder effect underlying many of their complex phenotypes, and the genetic basis for diseases such as inherited epilepsies could potentially be less complex in dogs than in humans (Ostrander and Kruglyak, 2000; Lindblad-Toh et al., 2005). A founder effect is the effect on the resulting gene pool that occurs when a new isolated population is founded by a small number of individuals possessing limited genetic variation relative to the larger population from which they have migrated. As such, there has been keen interest in utilizing the dog as a model of inherited epilepsy for this disease in humans.
Recent literature supports the hereditary basis for IE in many dog breeds, with a variety of genetic inheritance models proposed. Most IEs in dogs are thought to be genetically based, and IE has been reported in nearly every dog breed as well as in mixed-breed dogs (Ekenstedt et al., 2012). The prevalence of epilepsy in certain breeds of dog has been documented to be much higher than the estimated 0.5 to 6% in the general population; for example, in the Belgian shepherd Tervueren and Groenendael variants, the prevalence was estimated in one study to be 9.5%, and as high as 33% in one extended Belgian shepherd family (Berendt et al., 2008, 2009). A prevalence of 18.3% was documented in Irish wolfhounds with IE, and a recent study of Petit Basset Griffon Vendeen dogs with IE revealed a prevalence of 8.9% (Casal et al., 2006; Gullov et al., 2011).
A genetic or familial hereditary basis for IE has been investigated and proposed for each of the breeds listed in Table 6.1, typically by describing the clinical phenotype, examining the pedigrees, and suggesting a potential mode of inheritance (Ekenstedt et al., 2012). Additional breeds, which include Australian shepherds, Norwich terriers, Italian spinoni and greater Swiss mountain dogs, are the subjects of current investigation into IE, but clinical descriptions and possible modes of inheritance for these breeds have not yet been published (Ekenstedt et al., 2012).
There may be a lack of sufficient information for other breeds suffering epilepsy to be definitively classified as having IE or even an inherited epilepsy syndrome; these include the boxer and Shetland sheepdog (Ekenstedt et al., 2012). Epilepsy has been reported in the boxer and was determined to have medium to high heritability but the dogs included in the study did not go through a thorough workup to rule out other causes of seizure activity, necessary for a diagnosis of idiopathic disease (Nielen et al., 2001). A report on epilepsy in the Shetland sheepdog indicated that it may be inherited in a multifactorial or autosomal dominant fashion but the dogs in the study had histopathological abnormalities in the brain calling the diagnosis into question (Morita et al., 2002).
There has been evidence for an autosomal recessive inheritance or a gene of major effect in the canine IE studies that used pedigree segregation analysis (see glossary), both of which could occur due to a founder effect. However, many studies have been unable to rule out polygenic inheritance. For example, in a study of IE in English springer spaniels, the mode of inheritance appeared to be partially penetrant autosomal recessive or polygenic (Patterson et al., 2005) Similarly, in Vizslas, segregation analysis showed that IE in this breed is likely inherited in an autosomal recessive manner, but polygenic inheritance could not be excluded (Patterson et al., 2003). A large family of standard poodles with ‘probable’ IE, exhibiting focal seizure activity, was evaluated using segregation analysis (Licht et al., 2007). The dogs did not undergo CSF analysis or MRI. The segregation analysis strongly suggested that IE in this breed is inherited as a simple recessive trait with complete or almost complete penetrance. Idiopathic epilepsy was diagnosed in 146 Irish wolfhounds in one study that went on to use segregation analysis to evaluate the mode of inheritance of the disease, but again IE was not confirmed with CSF analysis or MRI (Casal et al., 2006). Pedigree examination revealed several features consistent with an autosomal recessive mode of inheritance in this breed. A simple dominant mode of inheritance could be excluded because most affected dogs were born to clinically normal parents. Although an X-linked mode of inheritance was discussed because the incidence of seizures in males was significantly higher than in females, in an X-linked recessive trait the fathers of affected females must also be affected, which was not the case. The estimated segregation ratio in this study was not consistent with a fully penetrant simple autosomal recessive mode of inheritance. It was therefore concluded that seizures in the Irish wolfhounds are inherited as an incompletely penetrant recessive trait with a sex predilection for older males in all of the affected dogs (Casal et al., 2006).
One recent candidate gene study examined four dog breeds for association or linkage to microsatellite markers in genes already known to be involved in human or murine IE (Ekenstedt et al., 2011). This study examined 52 genes encoding mostly ion channels and neurotransmitters in beagles, greater Swiss mountain dogs, English springer spaniels and Vizslas and found no major associations or linkages to IE in any breed. Genome-wide linkage studies have been undertaken in several dog breeds. A recently published study utilized DNA from 366 Belgian shepherd dogs, 74 of which were epileptic, from two extended family pedigrees (Oberbauer et al., 2010). This genome-wide linkage scan used 410 microsatellite markers and identified six quantitative trait loci (QTLs) on four chromosomes that appeared to be tentatively linked to IE, with no LOD scores achieving classical significance in excess of 3.0. An earlier study by the same group that used fewer dogs and fewer markers also did not achieve significant LOD scores; they identified three genomic regions with nominal linkage to the epileptic phenotype (Oberbauer et al., 2003).
Idiopathic Epilepsy Seizure Disorders with Known Mutations
The Lagotto Romagnolo breed segregates a recessive benign familial juvenile epilepsy, which typically remits by 4 months of age (Jokinen et al., 2007). The genetic cause for this syndrome has recently been identified as a truncating mutation in LGI2, an orthologue of the human epilepsy gene LGI1 (Seppala et al., 2011). This first canine IE gene sheds new light on the mechanisms of neuronal synaptic network remodelling during development, and shows that there is time sensitivity in the expression of LGI1 and LGI2, though they act on some of the same receptors. The Lagotto Romagnolo IE mutation discovery is particularly important because it is the first known canine epilepsy mutation directly linked to remission, it is developmentally stage-specific, and it sheds light on a novel pathway resulting in epilepsy. The same group that identified the mutation of LGI2 in the Lagotto Romagnolo breed investigated the Belgian shepherds with IE dominated by focal seizures with or without secondary generalization (Seppala et al., 2012). A simple Mendelian inheritance pattern was previously documented in this breed and so further study was necessary using molecular genetic tools (Berendt et al., 2009). A genome-wide association study using Affymetrix 50K SNP arrays in 40 IE Belgian shepherd dogs and 44 controls mapped the epilepsy locus on CFA37. Fine mapping study defined an approximate 1 Mb region including 12 genes of which none are known epilepsy genes or encode ion channels (Seppala et al., 2012). Exonic sequencing was performed for two candidate genes, KLF7 and ADAM23. No variation was found in KLF7 but a highly associated non-synonymous variant, G1203A (R387H) was present in the ADAM23 gene. Homozygosity for a two-SNP haplotype within the ADAM23 gene conferred the highest risk for epilepsy. ADAM23 interacts with known epilepsy proteins LGI1 and LGI2 but the data suggest that the ADAM23 variant may be a polymorphism (Seppala et al., 2012).
The Effect of Genetics on Anti-epileptic Medication Response
Investigations into the genetics of AEM response and AEM resistance in canine IE are a recent evolution. The concept of individualized medicine, namely using an individual’s genotype to guide administration of drug regimens, has shown promise in relation to human epilepsy. A recent study identified polymorphisms in the cytochrome P450 gene, CYP2C9, and a sodium channel gene, SCN1A, which are associated with the effective dosage of the AEMs carbamazepine and phenytoin (Tate et al., 2005). In veterinary medicine, a study of epileptic Border collies examined the ABCB1 gene (also called the MDR1 or multidrug resistance 1 gene). Affected dogs in this breed are often poorly controlled with AEMs, and resistant epilepsy develops in up to 71% of the cases (Hulsmeyer et al., 2010). It was determined that a sequence variation in ABCB1 intron 1 is associated with drug responsiveness in this breed (Alves et al., 2011). Seizure control and ABCB1 mutation was also recently investigated in Australian shepherd dogs but did not reveal an association (Weissl et al., 2012).
Five genes suggestive of association with drug response were identified in another study, which pooled many breeds of epileptic dogs together and used a custom SNP Bead Chip containing 30 genes involved in drug metabolism, drug targeting and drug transport to identify those associated with phenobarbital drug response (Kennerly et al., 2009). Two of the genes were ion channels (KCNQ3 and SCN2A2) and one was a neurotransmitter receptor (GABRA2).
Genetics and Feline Epilepsy
A genetic basis for recurrent seizures has been reported only in a closed colony of laboratory cats (Kuwabara et al., 2010). The colony consisted of 166 cats, of which 23 cats (16 males and 7 females) had recurrent seizures. The breed of these cats was not specified. The age at the time of the first seizure ranged between 4 and 12 months. Fourteen (9 males and 5 females) of these 23 cats underwent diagnostic investigations. Repeated general physical and neurological examinations, and results of haematology, serum biochemistry, electrolytes, blood gas analysis, urinalysis, serological tests for major viruses and Toxoplasma gondii, 1.5 Tesla MRI of the brain and CSF analysis were all normal. Interictal scalp-EEG under sedation (medetomidine) and/or anaesthesia (inhalation of sevoflurane) showed frequent interictal discharges consisting of spikes and sharp waves. All cats had focal onset complex seizures followed by secondary generalization into tonicclonic seizure. Most seizures occurred during sleep and were characterized by gradual head turning, unilateral facial twitching, hypersalivation and circling followed by generalized tonic-clonic manifestations. Seizures occurred in 6 of 14 epileptic cats (43%) with a frequency ranged from 0.5 to 19 seizures/month during the 2-month continuous video monitoring. Based on pedigree analysis an autosomal recessive mode of inheritance was hypothesized. Inbreeding of the epileptic cats produced six presumed homozygotic kittens. None of these cats had any seizures at the time of the study, when they were aged 5 to 14 months.
In the clinical setting, the term IE should be used carefully in cats presenting with recurrent seizures as breed predisposition is currently unknown and it is difficult to demonstrate a genetic or familial basis for the seizures. Cats with structural brain diseases can present with a normal neurological examination interictally (Quesnel et al., 1997). All cats presenting with seizures should undergo a thorough investigation for underlying seizure aetiologies.
References
Alves, L., Hulsmeyer, V., Jaggy, A., Fischer, A., Leeb, T. and Drogemuller, M. (2011) Polymorphisms in the ABCB1 gene in phenobarbital responsive and resistant idiopathic epileptic Border Collies. Journal of Veterinary Internal Medicine, 25, 484–489.
Asadi-Pooya, A.A., Emami, M. and Sperling, M.R. (2013) A clinical study of syndromes of idiopathic (genetic) generalized epilepsy. Journal of Neurological Science 324, 113–117.
Austin, J.K. and Caplan, R. (2007) Behavioral and psychiatric comorbidities in pediatric epilepsy: toward an integrative model. Epilepsia 48, 1639–1651.
Barnes, H.L., Chrisman, C.L., Mariani, C.L., Sims, M. and Alleman, A.R. (2004) Clinical signs, underlying cause, and outcome in cats with seizures: 17 cases (1997-2002). Journal of the American Veterinary Medical Association 225, 1723–1726.
Baulac, S. and Baulac, M. (2009) Advances on the genetics of mendelian idiopathic epilepsies. Neurologic Clinics 27, 1041–61.
Berendt, M. and Gram, L. (1999) Epilepsy and seizure classification in 63 dogs: a reappraisal of veterinary epilepsy terminology. Journal of Veterinary Internal Medicine 13, 14–20.
Berendt, M., Gredal, H., Pedersen, L.G., Alban, L. and Alving, J. (2002) A cross-sectional study of epilepsy in Danish Labrador Retrievers: prevalence and selected risk factors. Journal of Veterinary Internal Medicine 16, 262–268.
Berendt, M., Gredal, H. and Alving, J. (2004) Characteristics and phenomenology of epileptic partial seizures in dogs: similarities with human seizure semiology. Epilepsy Research 61, 167–173.
Berendt, M., Gredal, H., Ersboll, A.K. and Alving, J. (2007) Premature death, risk factors, and life patterns in dogs with epilepsy. Journal of Veterinary Internal Medicine 21, 754–759.
Berendt, M., Gullov, C.H., Christensen, S.L., Gudmundsdottir, H., Gredal, H., Fredholm, M. and Alban, L. (2008) Prevalence and characteristics of epilepsy in the Belgian shepherd variants Groenendael and Tervueren born in Denmark 1995-(2004). Acta Veterinaria Scandinavica 50, 51.
Berendt, M., Gullov, C.H. and Fredholm, M. (2009) Focal epilepsy in the Belgian shepherd: evidence for simple Mendelian inheritance. Journal of Small Animal Practice 50, 655–661.
Berkovic, S.F. and Scheffer, I.E. (1997) Epilepsies with single gene inheritance. Brain and Development 19, 13–18.
Berkovic, S.F., Mcintosh, A., Howell, R.A., Mitchell, A., Sheffield, L.J. and Hopper, J.L. (1996) Familial temporal lobe epilepsy: a common disorder identified in twins. Annals of Neurology 40, 227–235.
Bielfelt, S.W., Redman, H.C. and Mcclellan, R.O. (1971) Sire- and sex-related differences in rates of epileptiform seizures in a purebred beagle dog colony. American Journal of Veterinary Research 32, 2039–2048.
Biervert, C., Schroeder, B.C., Kubisch, C., Berkovic, S.F., Propping, P., Jentsch, T.J. and Steinlein, O.K. (1998) A potassium channel mutation in neonatal human epilepsy. Science 279, 403–406.
Brauer, C., Kastner, S.B., Rohn, K., Schenk, H.C., Tunsmeyer, J. and Tipold, A. (2012) Electroencephalographic recordings in dogs suffering from idiopathic and symptomatic epilepsy: diagnostic value of interictal short time EEG protocols supplemented by two activation techniques. Veterinary Journal 193, 185–192.
Casal, M.L., Munuve, R.M., Janis, M.A., Werner, P. and Henthorn, P.S. (2006) Epilepsy in Irish Wolfhounds. Journal of Veterinary Internal Medicine 20, 131–135.
Dichter, M.A. (2009) Emerging concepts in the pathogenesis of epilepsy and epileptogenesis. Archives of Neurology 66, 443–447.
Donner, E.J. (2011) Explaining the unexplained; expecting the unexpected: where are we with sudden unexpected death in epilepsy? Epilepsy Currents 11, 45–49.
Ekenstedt, K.J. and Oberbauer, A.M. (2013) Inherited epilepsy in dogs. Top Companion Animal Medicine 28(2), 51–58.
Ekenstedt, K.J., Patterson, E.E., Minor, K.M. and Mickelson, J.R. (2011) Candidate genes for idiopathic epilepsy in four dog breeds. BMC Genetics 12, 38.
Ekenstedt, K.J., Patterson, E.E. and Mickelson, J.R. (2012) Canine epilepsy genetics. Mammalian Genome 23, 28–39.
Engelborghs, S., D’Hooge, R. and De Deyn, P.P. (2000) Pathophysiology of epilepsy. Acta Neurologica Belgica 100, 201–213.
Escayg, A., Macdonald, B.T., Meisler, M.H., Baulac, S., Huberfeld, G., An-Gourfinkel, I., Brice, A., Leguern, E., Moulard, B., Chaigne, D., Buresi, C. and Malafosse, A. (2000) Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nature Genetics 24, 343–345.
Falco, M.J., Barker, J. and Wallace, M.E. (1974) The genetics of epilepsy in the British Alsatian. Journal of Small Animal Practice 15, 685–692.
Famula, T.R., Oberbauer, A.M. and Brown, K.N. (1997) Heritability of epileptic seizures in the Belgian tervueren. Journal of Small Animal Practice 38, 349–352.
Ficker, D.M., So, E.L., Shen, W.K., Annegers, J.F., O’Brien, P.C., Cascino, G.D. and Belau, P.G. (1998) Population-based study of the incidence of sudden unexplained death in epilepsy. Neurology 51, 1270–1274.
Gaitatzis, A., Carroll, K., Majeed, A. and W. Sander, J. (2004) The epidemiology of the comorbidity of epilepsy in the general population. Epilepsia 45, 1613–1622.
Gardiner, M. (2005) Genetics of idiopathic generalized epilepsies. Epilepsia 46(Suppl. 9), 15–20.
Gardiner, R.M. (2000) Impact of our understanding of the genetic aetiology of epilepsy. Journal of Neurology 247, 327–334.
Gastens, A.M., Brandt, C., Bankstahl, J.P. and Loscher, W. (2008) Predictors of pharmacoresistant epilepsy: pharmacoresistant rats differ from pharmacoresponsive rats in behavioral and cognitive abnormalities associated with experimentally induced epilepsy. Epilepsia 49, 1759–1776.
Greenberg, D.A. and Pal, D.K. (2007) The state of the art in the genetic analysis of the epilepsies. Current Neurology and Neuroscience Reports 7, 320–328.
Gullov, C.H., Toft, N., Baadsager, M.M. and Berendt, M. (2011) Epilepsy in the Petit Basset Griffon Vendeen: prevalence, semiology, and clinical phenotype. Journal of Veterinary Internal Medicine 25, 1372–1378.
Gullov, C.H., Toft, N. and Berendt, M. (2012) A longitudinal study of survival in Belgian Shepherds with genetic epilepsy. Journal of Veterinary Internal Medicine 26, 1115–1120.
Gurnett, C.A. and Hedera, P. (2007) New ideas in epilepsy genetics: novel epilepsy genes, copy number alterations, and gene regulation. Archives of Neurology 64, 324–328.
Hall, S.J. and Wallace, M.E. (1996) Canine epilepsy: a genetic counselling programme for keeshonds. Veterinary Record 138, 358–360.
Hauser, W.A., Annegers, J.F. and Elveback, L.R. (1980) Mortality in patients with epilepsy. Epilepsia 21, 399–412.
Haut, S.R., Hall, C.B., Masur, J. and Lipton, R.B. (2007) Seizure occurrence: precipitants and prediction. Neurology 69, 1905–1910.
Heinrichs, S.C. and Seyfried, T.N. (2006) Behavioral seizure correlates in animal models of epilepsy: a road map for assay selection, data interpretation, and the search for causal mechanisms. Epilepsy and Behavior 8, 5–38.
Heynold, Y., Faissler, D., Steffen, F. and Jaggy, A. (1997) Clinical, epidemiological and treatment results of idiopathic epilepsy in 54 labrador retrievers: a long-term study. Journal of Small Animal Practice 38, 7–14.
Holliday, T.A., Cunningham, J.G. and Gutnick, M.J. (1970) Comparative clinical and electroencephalographic studies of canine epilepsy. Epilepsia 11, 281–292.
Hulsmeyer, V., Zimmermann, R., Brauer, C., Sauter-Louis, C. and Fischer, A. (2010) Epilepsy in Border Collies: clinical manifestation, outcome, and mode of inheritance. Journal of Veterinary Internal Medicine 24, 171–178.
Imbrici, P., Jaffe, S.L., Eunson, L.H., Davies, N.P., Herd, C., Robertson, R., Kullmann, D.M. and Hanna, M.G. (2004) Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain 127, 2682–2692.
Jaggy, A. and Bernardini, M. (1998) Idiopathic epilepsy in 125 dogs: a long-term study. Clinical and electroencephalographic findings. Journal of Small Animal Practice 39, 23–29.
Jaggy, A., Faissler, D., Gaillard, C., Srenk, P. and Graber, H. (1998) Genetic aspects of idiopathic epilepsy in Labrador retrievers. Journal of Small Animal Practice 39, 275–280.
Jeserevics, J., Viitmaa, R., Cizinauskas, S., Sainio, K., Jokinen, T.S., Snellman, M., Bellino, C. and Bergamasco, L. (2007) Electroencephalography findings in healthy and Finnish Spitz dogs with epilepsy: visual and background quantitative analysis. Journal of Veterinary Internal Medicine 21, 1299–1306.
Jokinen, T.S., Metsahonkala, L., Bergamasco, L., Viitmaa, R., Syrja, P., Lohi, H., Snellman, M., Jeserevics, J. and Cizinauskas, S. (2007) Benign familial juvenile epilepsy in Lagotto Romagnolo dogs. Journal of Veterinary Internal Medicine 21, 464–471.
Kanner, A.M. (2006) Depression and epilepsy: a new perspective on two closely related disorders. Epilepsy Currents 6, 141–146.
Kathmann, I., Jaggy, A., Busato, A., Bartschi, M. and Gaillard, C. (1999) Clinical and genetic investigations of idiopathic epilepsy in the Bernese mountain dog. Journal of Small Animal Practice 40, 319–325.
Kearsley-Fleet, L., O’Neill, D.G., Volk, H.A., Church, D.B., Brodbelt, D.C. (2013). Prevalence and risk factors for canine epilepsy of unknown origin in the uK. Veterinary Record 30, 172(13), 338.
Kennerly, E.M., Idaghdour, Y., Olby, N.J., Munana, K.R. and Gibson, G. (2009) Pharmacogenetic association study of 30 genes with phenobarbital drug response in epileptic dogs. Pharmacogenetics and Genomics 19, 911–922.
Koskinen, L.L.E., Seppala, E.H. and Virta, M. (2012) Genetics of idiopathic epilepsy in the schipperke dog breed. Proceedings of the 6th International Conference on Advances in Canine and Feline Genomics and Inherited Diseases.
Kuwabara, T., Hasegawa, D., Ogawa, F., Kobayashi, M., Fujita, M., Suzuki, H., Matsuki, N. and Orima, H. (2010) A familial spontaneous epileptic feline strain: a novel model of idiopathic/genetic epilepsy. Epilepsy Research 92, 85–88.
LaFrance, W.C., Jr, Kanner, A.M. and Hermann, B. (2008) Psychiatric comorbidities in epilepsy. International Review of Neurobiology 83, 347–383.
Licht, B.G., Licht, M.H., Harper, K.M., Lin, S., Curtin, J.J., Hyson, L.L. and Willard, K. (2002) Clinical presentations of naturally occurring canine seizures: similarities to human seizures. Epilepsy and Behavior 3, 460–470.
Licht, B.G., Lin, S., Luo, Y., Hyson, L.L., Licht, M.H., Harper, K.M., Sullivan, S.A., Fernandez, S.A. and Johnston, E.V. (2007) Clinical characteristics and mode of inheritance of familial focal seizures in Standard Poodles. Journal of the American Veterinary Medical Association 231, 1520–1528.
Lindblad-Toh, K., Wade, C.M., Mikkelsen, T.S., Karlsson, E.K., Jaffe, D.B., Kamal, M., Clamp, M., Chang, J.L., Kulbokas, E.J., 3rd, Zody, M.C., Mauceli, E., Xie, X., Breen, M. et al. (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819.
McNamara, J.O. (1999) Emerging insights into the genesis of epilepsy. Nature 399, A15–22.
Michelucci, R., Passarelli, D., Pitzalis, S., Dal Corso, G., Tassinari, C.A. and Nobile, C. (2000) Autosomal dominant partial epilepsy with auditory features: description of a new family. Epilepsia 41, 967–970.
Monteiro, R., Adams, V., Keys, D. and Platt, S.R. (2012) Canine idiopathic epilepsy: prevalence, risk factors and outcome associated with cluster seizures and status epilepticus. Journal of Small Animal Practice 53, 526–530.
Morita, T., Shimada, A., Takeuchi, T., Hikasa, Y., Sawada, M., Ohiwa, S., Takahashi, M., Kubo, N., Shibahara, T., Miyata, H. and Ohama, E. (2002) Cliniconeuropathologic findings of familial frontal lobe epilepsy in Shetland sheepdogs. Canadian Journal of Veterinary Research 66, 35–41.
Neligan, A., Bell, G.S., Johnson, A.L., Goodridge, D.M., Shorvon, S.D. and Sander, J.W. (2011) The long-term risk of premature mortality in people with epilepsy. Brain 134, 388–395.
Nielen, A.L., Janss, L.L. and Knol, B.W. (2001) Heritability estimations for diseases, coat color, body weight, and height in a birth cohort of Boxers. American Journal of Veterinary Research 62, 1198–1206.
Nuyen, J., Schellevis, F.G., Satariano, W.A., Spreeuwenberg, P.M., Birkner, M.D., Van Den Bos, G.A. and Groenewegen, P.P. (2006) Comorbidity was associated with neurologic and psychiatric diseases: a general practice-based controlled study. Journal of Clinical Epidemiology 59, 1274–1284.
Oberbauer, A.M., Grossman, D.I., Irion, D.N., Schaffer, A.L., Eggleston, M.L. and Famula, T.R. (2003) The genetics of epilepsy in the Belgian tervuren and sheepdog. Journal of Heredity 94, 57–63.
Oberbauer, A.M., Belanger, J.M., Grossman, D.I., Regan, K.R. and Famula, T.R. (2010) Genome-wide linkage scan for loci associated with epilepsy in Belgian shepherd dogs. BMC Genetics 11, 35.
Ostrander, E.A. and Kruglyak, L. (2000) unleashing the canine genome. Genome Research 10, 1271–1274.
Pákozdy, A., Leschnik, M., Tichy, A.G. and Thalhammer, J.G. (2008) Retrospective clinical comparison of idiopathic versus symptomatic epilepsy in 240 dogs with seizures. Acta Veterinaria Hungarica 56, 471–483.
Patterson, E.E., Mickelson, J.R., Da, Y., Roberts, M.C., Mcvey, A.S., O’Brien, D.P., Johnson, G.S. and Armstrong, P.J. (2003) Clinical characteristics and inheritance of idiopathic epilepsy in Vizslas. Journal of Veterinary Internal Medicine 17, 319–325.
Patterson, E.E., Armstrong, P.J., O’Brien, D.P., Roberts, M.C., Johnson, G.S. and Mickelson, J.R. (2005) Clinical description and mode of inheritance of idiopathic epilepsy in English springer spaniels. Journal of the American Veterinary Medical Association 226, 54–58.
Podell, M., Fenner, W.R. and Powers, J.D. (1995) Seizure classification in dogs from a nonreferral-based population. Journal of the American Veterinary Medical Association 206, 1721–1728.
Proschowsky, H.F., Rugbjerg, H. and Ersboll, A.K. (2003) Mortality of purebred and mixed-breed dogs in Denmark. Preventive Veterinary Medicine 58, 63–74.
Quesnel, A.D., Parent, J.M., Mcdonell, W., Percy, D. and Lumsden, J.H. (1997) Diagnostic evaluation of cats with seizure disorders: 30 cases (1991-1993). Journal of the American Veterinary Medical Association 210, 65–71.
Schriefl, S., Steinberg, T.A., Matiasek, K., Ossig, A., Fenske, N. and Fischer, A. (2008) Etiologic classification of seizures, signalment, clinical signs, and outcome in cats with seizure disorders: 91 cases (2000)-(2004). Journal of the American Veterinary Medical Association 233, 1591–1597.
Seppala, E.H., Jokinen, T.S., Fukata, M., Fukata, Y., Webster, M.T., Karlsson, E.K., Kilpinen, S.K., Steffen, F., Dietschi, E., Leeb, T., Eklund, R., Zhao, X., Rilstone, J.J., Lindblad-Toh, K., Minassian, B.A. and Lohi, H. (2011) LGI2 truncation causes a remitting focal epilepsy in dogs. PLoS Genetics 7, e1002194.
Seppala, E.H., Koskinen, L.L., Gullov, C.H., Jokinen, P., Karlskov-Mortensen, P., Bergamasco, L., Baranowska Korberg, I., Cizinauskas, S., Oberbauer, A.M., Berendt, M., Fredholm, M. and Lohi, H. (2012) Identification of a novel idiopathic epilepsy locus in Belgian Shepherd dogs. PLoS One 7, e33549.
Shihab, N., Bowen, J. and Volk, H.A. (2011) Behavioral changes in dogs associated with the development of idiopathic epilepsy. Epilepsy and Behavior 21, 160–167.
Srenk, P. and Jaggy, A. (1996) Interictal electroencephalographic findings in a family of golden retrievers with idiopathic epilepsy. Journal of Small Animal Practice 37, 317–321.
Steinlein, O.K. (1998) New insights into the molecular and genetic mechanisms underlying idiopathic epilepsies. Clinical Genetics 54, 169–175.
Steinlein, O.K., Mulley, J.C., Propping, P., Wallace, R.H., Phillips, H.A., Sutherland, G.R., Scheffer, I.E. and Berkovic, S.F. (1995) A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nature Genetics 11, 201–203.
Surges, R., Thijs, R.D., Tan, H.L. and Sander, J.W. (2009) Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nature Reviews - Neurology 5, 492–504.
Tate, S.K., Depondt, C., Sisodiya, S.M., Cavalleri, G.L., Schorge, S., Soranzo, N., Thom, M., Sen, A., Shorvon, S.D., Sander, J.W., Wood, N.W. and Goldstein, D.B. (2005) Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin. Proceedings of the National Academy of Sciences USA 102, 5507–5512.
Tellez-Zenteno, J.F., Matijevic, S. and Wiebe, S. (2005) Somatic comorbidity of epilepsy in the general population in Canada. Epilepsia 46, 1955–1962.
Thomas, W.B. (2000) Idiopathic epilepsy in dogs. Veterinary Clinics of North America - Small Animal Practice 30, 183–206, vii.
Van Gelder, N.M., Janjua, N.A., Metrakos, K., Macgibbon, B. and Metrakos, J.D. (1980) Plasma amino acids in 3/sec spike-wave epilepsy. Neurochemical Research 5, 659–671.
Viitmaa, R., Cizinauskas, S., Bergamasco, L.A., Kuusela, E., Pascoe, P., Teppo, A.M., Jokinen, T.S., Kivisaari, L. and Snellman, M. (2006) Magnetic resonance imaging findings in Finnish Spitz dogs with focal epilepsy. Journal of Veterinary Internal Medicine 20, 305–310.
Viitmaa, R., Cizinauskas, S., Orro, T., Niilo-Rämä, M., Gordin, E., Lohi, H., Seppälä, E.H., Bragge, H. and Snellman, M. (2013) Phenotype, inheritance characteristics, and risk factors for idiopathic epilepsy in Finnish Spitz dogs. Journal of the American Veterinary Medical Association 243(7), 1001–1009.
S. Platt and L. De Risio
Wallace, R.H., Wang, D.W., Singh, R., Scheffer, I.E., George, A.L., Jr, Phillips, H.A., Saar, K., Reis, A., Johnson, E.W., Sutherland, G.R., Berkovic, S.F. and Mulley, J.C. (1998) Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nature Genetics 19, 366–370.
Weissl, J., Hulsmeyer, V., Brauer, C., Tipold, A., Koskinen, L.L., Kyostila, K., Lohi, H., Sauter-Louis, C., Wolf, M. and Fischer, A. (2012) Disease progression and treatment response of idiopathic epilepsy in Australian Shepherd dogs. Journal of Veterinary Internal Medicine 26, 116–125.
Winawer, M.R., Ottman, R., Hauser, W.A. and Pedley, T.A. (2000) Autosomal dominant partial epilepsy with auditory features: defining the phenotype. Neurology 54, 2173–2176.
Zuberi, S.M., Eunson, L.H., Spauschus, A., De Silva, R., Tolmie, J., Wood, N.W., Mcwilliam, R.C., Stephenson, J.B., Kullmann, D.M. and Hanna, M.G. (1999) A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 122(5), 817–825.


Plate 1. Copper-coloured iris in a 1-year 9-month-old female spayed domestic short hair cat with hepatic encephalopathy secondary to congenital extrahepatic portosystemic shunt (eye on the right) compared with the green iris of a normal littermate (eye on the left) (courtesy of Kelly Bowlt ©). Plate 2. Histology of the brain of a domestic short hair cat with thiamine (vitamin B1) deficiency. Note the focal haemorrhagic necrotic lesions localized to the lateral geniculate nuclei (courtesy of Professor Kaspar Matiasek ©).

Plate 3. Transverse section of the brain of a dog at the level of the caudate nuclei. Note the unilateral juxtacapsular lacunar ischaemic infarct (frame) (courtesy of Professor Kaspar Matiasek ©). Plate 4. Anterior uveitis in 12-month-old Persian cat with FIP (courtesy of Jamie Oliver ©).
5

6
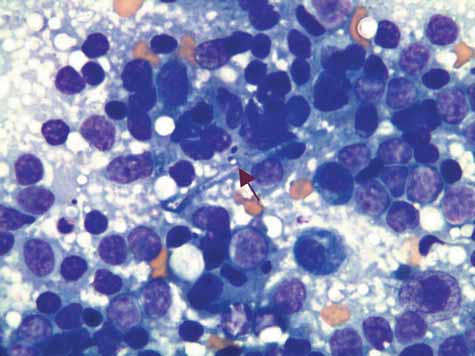
Plate 5. Transverse T1-weighted post-contrast MRI at the level of the frontal lobe of the dog in Fig. 5.10 demonstrates marked homogenous contrast uptake by the lesion, which exhibits well-defined borders within the parenchyma of the brain. Plate 6. A cytological impression smear of the lesion taken at the time of surgical biopsy of the dog in Fig. 5.10, demonstrates a marked mononuclear cell response surrounding a hyphae structure (arrow) compatible with a fungal disease. Culture of the tissue confirmed Aspergillus spp. infection.

Plate 7. Transverse section of the brain of the dog described in Fig. 5.11 at the level of the haemorrhagic tract within the left thalamus seen on MRI. Note the multiple haemorrhagic lesions (infarcts/necroses) in the cerebral cortex, subcortical white matter, right hippocampus and thalamus. Histology of the lesions is consistent with parasitic migrating ducts inciting vascular lesions. Plate 8. High-power field image of the thalamic lesion shown in Plate 7 shows intralesional vessel walls undergoing dystrophic mineralization and pigment cell aggregates (courtesy of Professor Kaspar Matiasek ©). Plate 9. Histology of the frontal lobe of a 4-year-old female husky with GME. Diffuse angiocentric cell infiltrates are wide-spread throughout white and grey matter of the frontal lobe. Perivascular epitheloid cells are evident at higher magnification (courtesy of Professor Kaspar Matiasek ©).
10a

10b
Caudal transtentorial herniation

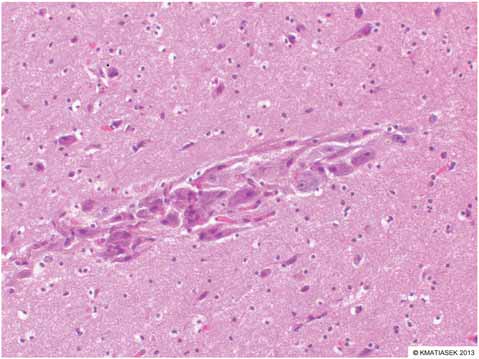

Plate 11. Nodular grey matter ectopia in the frontal lobe of a 2-year-old mixed breed dog with pharmacoresistant epilepsy (courtesy of Professor Kaspar Matiasek ©). Plate 12. Dysmorphic cortical neuron (arrow) in the frontal cortex of a 1-year-old dog with focal cortical dysplasia (courtesy of Professor Kaspar Matiasek ©).
13
14

15



Plate 17. Gross appearance of the right frontoparietal mass of the cat described in Fig. 5.18 following surgical resection. Histology of this mass is shown in Plate 18 (courtesy of Professor Kaspar Matiasek ©). Plate 18. Histology of the cat described in Fig. 5.18 confirmed the diagnosis of fibroblastic meningioma with cholesterol clefts (courtesy of Professor Kaspar Matiasek ©).



Plate 21. Mitochondrial encephalopathy in a Yorkshire terrier. Transverse section of the cerebrum shows characteristic bilateral and symmetrical necrotic areas in the thalamus (dashed line) and cerebral cortex, surrounding the tips of the sulci (arrows) (courtesy of Professor Kaspar Matiasek ©). Plate 22. Limbic encephalitis: prosencephalic transverse section of a feline brain with limbic encephalitis. Note the disruption and pallor of the hippocampus (HC) (courtesy of Professor Kaspar Matiasek ©).
Ascending neural radiations to cerebral cortex
Neural input
CN I Cerebral cortex Corpus callosum Thalamus Cerebellum Reticular formation

Spinal cord somatosensory pathways
Brainstem

Lateral geniculate
Occipital cortex
Sagittal view
Optic radiation, contralateral Occipital cortex, contraleteral
Lateral geniculate nucleus, contralateral Thalamus, contralateral Optic tract, contralateral Optic Chiasm
Plate 23. Schematic illustration of the ascending reticular activating system (ARAS), a complex neural network in the central core of the brainstem extending from the medulla oblongata through the pons and midbrain to the caudal diencephalon. The ARAS receives input from most sensory modalities via the spinal cord somatosensory pathways and the sensory component of several cranial nerves and projects diffusely, to the cerebral cortex to maintain a normal level of consciousness. Plate 24. Schematic illustration of the visual pathways. The visual stimulus is detected by the retina, the resulting impulse travels through the optic nerve (CN II) and optic chiasm where the majority of fibres (65–75%) in dogs and cats decussate, and continues through the optic tract to the lateral geniculate nucleus of the thalamus. From here the stimulus travels through the optic radiation to the occipital cortex. A lesion affecting any part of this pathway may result in impaired conscious vision.
| 25 | Dorsal view | 26 | |||||
|---|---|---|---|---|---|---|---|
| 10 | 11 | 1 | 2 | 3 | 4 | 6 | |
| 5 | |||||||
| 8 | 7 | ||||||
PUPPILLARY LIGHT REFLEX
Dorsal view
Retina Optic nerve Optic Chiasm Optic tract Parasympathetic
branch of CN III Pretectal nucleus in most rostal
part of midbrain (rostral colliculus) Parasympathetic
nucleus of CN III
Sagittal view
Pretectal nucleus contralateral
| 9 | |||||||
|---|---|---|---|---|---|---|---|
| Sagittal view | |||||||
| 6 | 5 | ||||||
| 8 | |||||||
| 4 | 1 Retina | Oculomotor nucleus | |||||
| 1 11 | 2 | 3 10 | 7 | 9 | 2 Optic nerve 3 Optic chiasm 4 Lateral geniculate nucleus 5 Occipital cortex 6 Motor cortex | Oculomotor nerve Optic chiasm | |
| 7 Pontine nucleus | |||||||
| 8 Cerebellar cortex | |||||||
| 9 Facial nucleus | |||||||
| 10 Facial nerve (CN VII) | |||||||
| 11 Orbicularis oculi | |||||||
Plate 25. Schematic illustration of the menace response pathway. The menacing stimulus is detected by the retina (1) and the resulting impulse travels through the optic nerve (2), optic chiasm (3) and the contralateral optic tract. The stimulus is relayed to the lateral geniculate nucleus (4) in the thalamus, travels through the optic radiation and synapses in the occipital cortex (5). The impulse travels rostrally to the motor cortex (6) and then through the internal capsule, crus cerebri and longitudinal fibres of the pons and synapses in the pontine nucleus (7). It then proceeds within the transverse fibres of the pons, through the middle cerebellar peduncle and synapses in the cerebellar cortex (8). The signal travels through the efferent cerebellar pathway and synapses on both facial nuclei (9). The impulse travels through the facial nerve (10) resulting in contraction of the orbicularis oculi muscle (11) and eyelid closure. A lesion affecting any part of this pathway may result in a menace response deficit.
Plate 26. Schematic illustration of the pupillary light reflex (PLR) pathway. A bright light stimulus is detected by the retina, the resulting impulse travels through the optic nerve (CN II), optic chiasm and optic tract. The stimulus is relayed to the pretectal nucleus within the rostral colliculus and from here to the parasympathetic nucleus of the oculomotor nerve (CN III). The stimulus then travels along the parasympathetic component of CN III to the iris sphincter muscle resulting in its contraction and constriction of the pupil. A lesion in any part of this pathway can affect the PLR.


Plate 27. Schematic illustration of the regions of the central and peripheral nervous system in which the lesion can be localized by means of the neurological examination. Plate 28. Post-processing analysis of a normal transverse canine brain following diffusion tensor imaging reveals the pathways of the white matter tracts in the corpus collosum. The colour differences represent different directions of the tracts.

Plate 30. CSF neutrophilic pleocytosis in an 8-month-old Beagle with spinal cervical hyperalgesia due to steroid-responsive meningitis-arteritis. Magnification 50×. (Courtesy of Fancesco Cian.)


7 Epidemiology of Canine Seizures
Simon Platt Bvm&S mrCvS, Dipl. ACvim (Neurology) Dipl. ECvN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
The prevalence of epilepsy in dogs has been estimated at 1% to 5% (Holliday et al., 1970; Podell et al., 1995). Epilepsy was confirmed in 711 of 90,004 (<1%) dogs examined at a veterinary teaching hospital in one study, and was significantly more common in purebreds (Bellumori et al., 2013). In the same study, 27,254 dogs had evidence of at least one genetic condition and of these epilepsy represented 2.6% (Bellumori et al., 2013). When just focusing on neurological referral cases, a study documented that the most frequent neurological localization was the forebrain affecting 908 of 4497 dogs admitted to the hospital (20%) (Fluehmann et al., 2006). Among the dogs with forebrain disease, idiopathic epilepsy was the most frequent diagnosis, which was found in 372 dogs (41%; 8% of all neurological referrals) (Fluehmann et al., 2006).
Prevalence of Canine Seizures Based on Semiological Classification
Specific seizure types have been associated with specific disease processes in humans; however, this still needs further evaluation in veterinary medicine.
In the past, generalized tonic-clonic seizures were considered the most common type of seizure in dogs with idiopathic epilepsy, and it was even claimed focal-onset seizures were inconsistent with a diagnosis of idiopathic epilepsy. However, more recent observations reveal this is clearly not the case and dogs with idiopathic epilepsy can have a variety of focal-onset seizures, including secondarily generalized seizures, with some individuals having more than one type of seizure (Heynold et al., 1997; Jaggy and Bernardini, 1998; Patterson et al., 2003, 2005; Pákozdy et al., 2008). Seventy dogs, representing 38 different breeds, with focal seizures were evaluated by Berendt and others in 2004 (Berendt et al., 2004). Sixty-one dogs (87%) had focal seizures with secondary generalization. In 16 of these dogs, seizure activity did on some occasions remain focal;
i.e. secondary generalization was not a constant finding in these dogs. Nine dogs (13%) had focal seizures without secondary generalization. Motor signs were reported in 48 dogs (69%). Autonomic signs were reported in 16 dogs (23%). Paroxysms of behavioural signs were reported in 56 dogs (80%). The signs reported lasted from 15 s to 3 min (Berendt et al., 2004). In 38 dogs (55%) both motor and behavioural signs were present during seizures. Nine dogs (13%) had seizures with both autonomic and behavioural signs. Seven dogs (10%) had seizures with both autonomic and motor signs. Four dogs (6%) had motor signs, autonomic signs and paroxysms
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
of behavioural signs in combination during seizures (Berendt et al., 2004).
A higher probability of symptomatic epilepsy has been documented in dogs with a short interictal interval and focal seizures, which supported the belief that focal seizures are an indication of a structural cerebral lesion (Podell et al., 1995; March, 1998; Licht et al., 2002). Similar results were reported in a later study by Berendt and Gram (Berendt and Gram, 1999). A more recent study of vizslas with inherited idiopathic epilepsy showed that focal seizures of variable frequency were the predominant seizure type (Patterson et al., 2003). Further studies demonstrated similar associations (Heynold et al., 1997; Licht et al., 2007; Hulsmeyer et al., 2010). The most recent evaluation of seizures in young dogs revealed that 67% of dogs less than 1 year of age with focal seizures were diagnosed with idiopathic epilepsy (IE) (Arrol et al., 2012). In this study of seizures in 136 dogs less than 1 year of age, seizures were classified as primarily focal in 39 (27.7%) dogs and generalized in 97 (71.3%) dogs. Focal seizures were present in 25.5% of the IE group, 34.8% of the structural group, 33% of the reactive group and 100% of the cryptogenic group. There was no relationship between the occurrence of focal seizures and the underlying aetiology.
Multiple studies have investigated IE in specific breeds and described the seizure types identified. Jaggy and others (1998) investigated Labrador retrievers with IE and found that 96% (53/55) of dogs showed generalized seizures, 89% with tonic-clonic motor activity with loss of consciousness; the remaining 11% had retained consciousness during the seizure (Jaggy et al., 1998). Two dogs (4%) had what were described as simple focal (partial) seizures. However, approximately two-thirds of the dogs had a prodromal phase, mainly characterized by restlessness and fear lasting for several hours. In these dogs, this episode was followed by a very short aura starting with unilateral focal motor activity of the head and then all the spinal muscles. Therefore, it is likely that the majority of the dogs in this study actually expressed focal seizures, which progressed to generalized activity (Jaggy et al., 1998). A smaller study of Danish Labrador retrievers found that 24% were classified with primary generalized seizures, whereas 12 dogs (70%) were classified with focal (partial) seizures (Berendt et al., 2002). The type of seizures could not be classified in one dog (6%). In the group experiencing focal seizures, two dogs (17%) experienced focal seizures alone (one with motor signs and one with autonomic signs), whereas ten dogs (83%) experienced focal seizures with secondary generalization (Berendt et al., 2002). In a study of 49 Border collies with IE, 28 dogs (57%) had pre-ictal symptoms such as vomiting, aggressive behaviour, salivation, seeking the owner’s attention or restlessness; these were normally observed up to 30 min before seizure onset (Hulsmeyer et al., 2010). In 18% (5/28) of dogs, owners recognized pre-ictal signs 1–2 days before seizure onset, consisting of lameness of one forelimb and decreased reaction to known commands. In all 49 dogs, the predominant seizure type was generalized. A focal onset was observed in 38 dogs (78%), mostly characterized by staring into space for seconds and lateral head turn or opisthotonus (focal onset seizure) (Hulsmeyer et al., 2010). In four dogs (8%), seizures were classified as primarily generalized because of absence of any initial motor, behavioural, or autonomic signs. In seven dogs (14%), seizures remained unclassified, as seizure onset had not been observed. Generalization was dominated by lateral posture, loss of consciousness, tonic-clonic movements of the limbs, salivation, repetitive jaw movements and urination. Twenty-two dogs (45%) occasionally had isolated focal motor seizures without secondary generalization, usually manifested as sudden uncontrolled head or face twitching mostly associated with impaired consciousness (which the authors referred to as complex focal seizures) (Hulsmeyer et al., 2010). Fifty Australian shepherds were evaluated over a 6-year period at multiple European centres (Weissl et al., 2012). Fifty-two per cent (26/50) of all dogs demonstrated focal seizures in addition to generalized seizures. Generalization was expressed in lateral posture, tonic-clonic limb movements, salivation and, occasionally, repetitive jaw movements, enuresis and vocalization. Focal seizures or focal onsets presented with focal tremors, salivation, dilated pupils and lateral head turn. Concomitant or solitary episodes with variable states of awareness and behavioural changes like panic attacks, sporadic aggressiveness, pacing, staring, or adverse reactions were common (Weissl et al., 2012). Of the 29 standard poodles with IE evaluated by Licht et al. (2007), 27 (93%) had evidence of focal onset seizures (Licht et al., 2007). Of these 27 dogs, nine (33%) had had at least one seizure that secondarily generalized. Thus, 11 of the 29 (38%) dogs had had at least one generalized seizure (either primary or secondarily generalized). In six of these 11 dogs, generalized seizures included tonic (stiffness and rigidity) and clonic (shaking, jerking or paddling) signs, and in four, generalized seizures included only clonic signs. For the remaining dog, the owner observed only tonic signs but did not see the seizures from the beginning. Of the 18 dogs that had focal seizures without secondary generalization, six had simple focal seizures (i.e. consciousness was preserved) and 12 had complex focal seizures (i.e. consciousness was impaired). Of the nine dogs that had focal onset seizures that secondarily generalized, eight had evidence of impaired consciousness during the focal onset. Autonomic signs included drooling (n = 11), panting (4), urination (1) and increased heart rate (1). Thirteen dogs had psychic signs (anxiety) and three had automatisms (two with licking, lip smacking or swallowing and one with circling) (Licht et al., 2002).
Prevalence of Canine Seizures Based on Aetiological Classification
idiopathic epilepsy
Various studies have documented the prevalence of IE as compared to the other aetiological classifications. A study of dogs less than 1 year of age revealed that IE was seen in 75% of cases (Arrol et al., 2012). In the same study, structural epilepsy comprised 17%, reactive 7% and cryptogenic less than 2% of the cases (Table 7.1).
Table 7.1. Seizure aetiology and associated outcome.
| Cause of seizures | Number of dogs affected | Outcome |
| Idiopathic | 102 (75%) | Known in 80 |
| 50 (62.5%) alive | ||
| 25 (31.3%) euthanasia due to | ||
| seizures | ||
| 5 (6.3%) euthanasia/death | ||
| unrelated to seizures | ||
| Symptomatic | 23 (16.9%) | Known in 19 |
| Hydrocephalus (6), inflammatory brain | 6 (31.6%) alive | |
| disease (6), infarct (3), degenerative | 12 (63.2%) euthanasia due | |
| disease (2), hydranencephaly (2), | to seizures | |
| neoplasia (2), canine distemper | 1 (5.3%) euthanasia/death | |
| virus (1), toxoplasmosis (1) and | unrelated to seizures | |
| L-2-hydroxyglutaric aciduria (1) | ||
| Probable | 2 (1.5%) | Known in 2 |
| symptomatic | 0 (0%) alive | |
| 2 (100%) euthanasia due to | ||
| seizures | ||
| 0 (0%) euthanasia/death | ||
| unrelated to seizures | ||
| Reactive | 9 (6.6%) | Known in 9 |
| Extrahepatic liver shunt (5), juvenile | 7 (77.8%) alive | |
| hypoglycaemia (2), intoxication (2) | 1 (11.1%) euthanasia due to seizures | |
| 1 (11.1%) euthanasia/death | ||
| unrelated to seizures |
The prevalence of IE in certain breeds of dog has been documented to be much higher than the estimated 0.5 to 5% in the general population; for example, in the Belgian shepherd Tervueren and Groenendael variants, the prevalence was estimated in one study to be 9.5%, and as high as 33% in one extended Belgian shepherd family (Berendt et al., 2008, 2009). A prevalence of 18.3% was documented in Irish wolfhounds with IE; recent studies of Petit Basset Griffon Vendeen dogs with IE revealed a prevalence of 8.9%, of Labrador retrievers in Switzerland revealed 6.9% and Danish Labrador retrievers were shown to have 3.1% prevalence (Jaggy et al., 1998; Berendt et al., 2002; Casal et al., 2006; Gullov et al., 2011).
Cryptogenic epilepsy
A recent study evaluated 214 client-owned dogs with an onset of epileptic seizures ³7 years of age (Schwartz et al., 2013). Forty-five (21%) dogs had a diagnosis of cryptogenic epilepsy and 169 (79%) had structural epilepsy. In dogs 7 to 9 years and ³10 years of age at the time of seizure onset, 31 of 106 (29%) and 14 of 108 (13%), respectively, had a diagnosis of cryptogenic epilepsy. Breeds of dogs with cryptogenic epilepsy appeared consistent with the overall dog population seen at the study hospital, apart from Siberian huskies. Even though numbers were small, this breed was apparently over-represented, compared with the percentage of Siberian huskies seen at that specific hospital during the study period (0.7%) (Schwartz et al., 2013). This may have been coincidental; alternatively, it may reflect a predisposition for dogs of this breed to develop epilepsy at an advanced age. At the last follow-up time of the study, most (40; 89%) dogs with cryptogenic epilepsy were receiving one or more anti-epileptic medication (AEM). Thirty-one of 37 (84%) dogs typically had one or more seizures per month following hospital discharge. Death was confirmed in 20 (44%) dogs with cryptogenic epilepsy and was related to seizures or AEMs in seven of them. Median survival time from onset of seizures was 52 months for all dogs with cryptogenic epilepsy. Median quality-of-life score (scale, 1 (poor) to 10 (excellent)) indicated by 34 owners of dogs with cryptogenic epilepsy was 10 before diagnosis and initiation of AEM treatment and 8 afterward (Schwartz et al., 2013).
Symptomatic (structural) epilepsy
Causes of recurrent epileptic seizures were analysed in 412 dogs referred to a veterinary hospital in Europe with epilepsy due to an intracranial disorder (i.e. not including reactive seizures) over an 11-year period (Steinmetz et al., 2013). The causes included head trauma (n = 64; 15.5%), brain tumours (n = 33; 8%), brain inflammation (n = 25; 6%), cortical dysgenesis (n = 9; 2%), and several other probable causes (n = 17; 4%), including scars (2), hydrocephalus (10), cysts (2) and vascular malformations (3) (Steinmetz et al., 2013). Furthermore, six dogs had a dual pathology (e.g. hydrocephalus and cysts) that most likely caused the structural epilepsy. In the majority (n = 258; 63%) of the 412 dogs, a cause could not be unequivocally identified, so these animals were assigned to the idiopathic/cryptogenic group, which was not further separated in this study. In an earlier study on 211 dogs with recurrent seizures referred to a veterinary hospital over a period of 5 years due to intracranial causes, only three (1.4%) had a history of head trauma but 18% had brain tumours and 11% had encephalitis (Pákozdy et al., 2008).
Inflammation
Seizures occur in about 13% of dogs with all causes of CNS inflammatory disease based on one large study (Tipold, 1995). Canine distemper virus (CDV) encephalitis is the most common infectious inflammatory cause of seizures in dogs younger than 1 year of age (Podell et al., 1995). Seizures often are focal, characterized by ‘chewing gum’ actions, or consist of generalized motor activity (Tipold, 1995). Similarly, seizures have been reported with post-vaccinal CDV encephalitis in puppies (Cornwell et al., 1988). These seizures are often progressive and refractory to anti-epileptic drug therapy. Identifying the underlying inflammatory disease process is important because the disease will continue to progress without appropriate treatment.
Dogs with non-infectious inflammatory disorders with cerebral cortical involvement frequently clinically manifest seizure activity. Granulomatous meningoencephalomyelitis (GME) is an inflammatory disease of the white matter of the brain. The disseminated form is usually acute and rapidly progressive, whereas the focal form progresses more slowly. Necrotizing meningoencephalitis (NME) is a non-suppurative inflammatory central nervous system (CNS) disease that is common in pugs, with variants recognized in other breeds such as the Maltese, Chihuahua, Yorkshire terrier, Pekingese and French bulldog. In pugs, NME lesions are most common in the cerebral cortex and subcortical white matter. A recent study including 60 pug dogs with necropsy confirmed NME-documented seizure activity in 100% (Levine et al., 2008).
Intracranial neoplasia
Epileptic seizures are a well-recognized manifestation of intracranial neoplasia in dogs (Bagley et al., 1999; Platt and Haag, 2002; Snyder et al., 2006; Schwartz et al., 2011). Not all dogs with brain tumours will develop seizures, hence certain factors must exist that promote epileptogenesis; however, the pathogenesis of tumour-associated seizures is poorly understood (Beaumont and Whittle, 2000; Schaller and Ruegg, 2003; Shamji et al., 2009). The vast majority of intracranial tumours originate from non-neuronal cells that lack intrinsic epileptogenic properties with the capability to generate action potentials; therefore, in most instances, seizure development must depend on the tumour’s effects on the adjacent neuronal tissue. Various pathophysiological mechanisms for brain tumour-associated epileptogenesis have been proposed, mainly relating to the peritumoural area. Proposed epileptogenic factors include: local cerebral ischaemia; morphological changes causing isolation and denervation of cerebral cortical areas; neuronal, axonal and synaptic plasticity; perturbation in balance of neurotransmitters and their respective receptors; ionic changes and alterations in pH; triggering of a peritumoural immune response and modified intercellular communication of surrounding glial cells (Beaumont and Whittle, 2000; Schaller and Ruegg, 2003). Generation of seizure activity likely results from interplay of these factors and additionally depends on the patients’ susceptibility to seizure development. The causal relationship between intracranial neoplasia in dogs and seizure activity has frequently been reported but studies identifying risk factors for seizure development are lacking (Bagley and Gavin, 1998; Bagley et al., 1999; Snyder et al., 2006). To identify clinical risk factors for seizures in dogs with intracranial neoplasia, a recent cross-sectional retrospective study was performed on 68 dogs with histopathologically confirmed primary or secondary intracranial neoplasia, complete clinical history and magnetic resonance imaging of the brain was conducted (Schwartz et al., 2011). Prevalence of findings was compared between dogs with and without seizures. Forty-two dogs had tumour-related seizures and the remaining 26 were seizure-free. Tumour types included meningioma (23 dogs with and five without seizures), glioma (no dogs with and six without seizures), choroid plexus tumour (two dogs without seizures), neuroblastoma (one dog with seizures) and metastatic/invasive tumours including lymphoma (nine dogs with and 13 without seizures). On the basis of multi-variable logistic regression analysis, risk factors for seizures associated with intracranial neoplasia were magnetic resonance imaging findings consistent with the presence of neoplastic tissue in frontal lobe, marked gadolinium enhancement and magnetic resonance imaging findings of subfalcine and/or subtentorial herniation. The proportion of dogs with seizure activity was highest among individuals with meningiomas followed by gliomas, and metastatic/invasive tumours, however, there was no significant relationship between tumour type and seizure development. A true difference may have remained undetected due to the limited number of dogs that fulfilled the inclusion criteria (Schwartz et al., 2011).
In humans, the occurrence of tumourassociated seizures is highly dependent on tumour type. Seizure frequency is highest in dysembryoblastic neuroepithelial tumours (DNETs); also, oligodendrogliomas and astrocytomas are reported to cause seizures in more than half of patients. Among these, especially slow-growing, low-grade gliomas are associated with seizure development (Beaumont and Whittle, 2000; van Breemen et al., 2007; Shamji et al., 2009). A single study on canine brain tumours could detect such an increased likelihood of seizures in a certain tumour type. Dogs with oligodendrogliomas were identified to more likely present with seizures when compared to dogs with other primary brain tumours (Snyder et al., 2006). The proportion of dogs with seizures was highest when the temporal, frontal or parietal lobes or the olfactory bulb contained neoplastic tissue, similarly to previous reports (Bagley et al., 1999). This is in accordance with the human literature that found highest occurrence rates when these regions were affected whereas involvement of the occipital lobe was less commonly associated with epileptogenesis (Mahaley and Dudka, 1981; Lieu and Howng, 2000; Liigant et al., 2001; Lynam et al., 2007). Dogs with primary or secondary intracranial neoplasia are at risk of seizures, particularly those with tumours that affect the frontal lobe, enhance markedly with gadolinium or cause subfalcine and/or subtentorial herniation. Two-thirds of the dogs included in this case series developed epileptic seizures as a clinical sign of intracranial neoplasia and in three-quarters of these dogs a seizure was the first clinical sign noted by the owners. These figures emphasize the high prevalence of tumour-associated epilepsy and the importance of intracranial neoplasia as a potential underlying cause for seizures. Other studies have found a lower prevalence of seizures in dogs with intracranial neoplasia. Snyder et al. (2006) found 51% to show seizure activity, whereas 45% of dogs presented by Bagley and others (1999) displayed tumour-associated seizures (Bagley et al., 1999; Snyder et al., 2006).
A human study found all tumour-associated seizures to start focally, of which half subsequently experienced generalization (Liigant et al., 2001). A similar situation could be expected in canine patients, nevertheless, focal seizures may remain undetected in many instances. In the presented study, seizure severity in more than half of the dogs with tumourassociated seizures had progressed to cluster seizures at the time of MR imaging.
Head trauma
Traumatic brain injury (TBI) is a major cause of epilepsy in people (Lowenstein, 2009). Epidemiologic human studies have found that post-traumatic epilepsy (PTE) accounts for approximately 20% of structural epilepsy in the general population and 5% of all patients seen at specialized epilepsy centres (Agrawal et al., 2006). The main, critical determinant of the development of PTE is the severity of the head injury (Lowenstein, 2009). Key risk factors include skull fracture, intracranial haematoma, and a depressed level of consciousness at the time of admission to the emergency department. In addition, the occurrence of seizures within the first week after TBI also appears to be a risk factor for subsequent development of epilepsy (Lowenstein, 2009).
The mechanisms underlying development of PTE are not completely understood (D’Ambrosio and Perucca, 2004; Pitkanen et al., 2009). Furthermore, there is a lack of reliable biomarkers that allow predicting which patients develop epilepsy after TBI (Lowenstein, 2009). There is often a delay of months to years in the emergence of epilepsy after the initial injury.
Of the 236 dogs with head injury in a large retrospective epidemiological study of dogs with epilepsy, 44 (18.6%) exhibited early and/or late seizures (Steinmetz et al., 2013). Observed seizure types were focal and generalized tonic-clonic; convulsive status epilepticus or cluster seizures were observed in four dogs. Age at head injury in these 44 dogs was
3.69 years (range 0.16–15 years). Twenty-five dogs were female, 13 male, four male-neutered and two spayed; the proportion of female dogs was significantly higher than the proportion of male animals. The predominant cause of head injury in these 44 dogs was a car accident (n = 20), followed by punches (9), falls (8), horse kicks (4) and bites (3). Of the 198 dogs that did not die immediately after or within 1 week following head injury, 13 dogs (6.6%) developed late recurrent seizures, indicating development of epilepsy. The average latency between head injury and onset of epilepsy was 1 year. Most dogs (86%) with head injury had closed injury, but 14% had open injury with a fractured skull and, in nine dogs, penetrating injury. Open injury was associated significantly more often with seizures (early or late) than closed injury. Furthermore, 14.3% of the dogs with open injury developed epilepsy compared to 5.3% of the dogs with closed injury, which, however, was not statistically significant. The average latency between head injury and onset of epilepsy was similar in dogs with closed and open head injuries. In dogs with penetrating injury, 44% showed early seizures after the injury, but none of the dogs developed epilepsy, which may be secondary to the fact that only seven dogs survived the first week after head injury.
A separate study evaluated 259 dogs admitted with head trauma over a 10-year period at The Ohio State University Veterinary Medical Center (Friedenberg et al., 2012). Overall, 3.5% of dogs with head trauma developed in-hospital seizures, and 6.8% of dogs with head trauma for which follow-up information was available developed seizures after hospital discharge, compared with an epilepsy rate of 1.4% in the authors’ hospital. Dogs that developed in-hospital seizures were significantly more likely to have been hit by a car or experienced acceleration-deceleration injury. Additionally, 10% of dogs with traumatic brain injury had in-hospital seizures. No visit or patient characteristics were significantly associated with the development of out-of-hospital seizures (Friedenberg et al., 2012).
Vascular disease
Stroke is increasingly identified as the underlying cause of acute neurological disease in dogs by magnetic resonance imaging (MRI) or computed tomography (CT) (Platt and Garosi, 2003; Garosi and McConnell, 2005; Garosi et al., 2005, 2006; Platt et al., 2006; Garosi, 2010; Goncalves et al., 2011). A stroke can be caused by either a haemorrhage in the brain,
i.e. haemorrhagic stroke, or by thromboembolic occlusion of a cerebral artery causing ischaemic stroke (Wessmann et al., 2009). In dogs, the majority of strokes appear to be ischaemic (Wessmann et al., 2009). The typical clinical picture in dogs with ischaemic stroke is characterized by acute neurological deficits, varying according to the site and the extension of the lesion (Garosi et al., 2006; Garosi, 2010; Goncalves et al., 2011). Neurological signs include altered mentation, hemi- or tetraparesis, circling, cranial nerve deficits and vestibular signs. Deterioration can be seen within the first 24 h of the insult due to progressive oedema, but should generally not be expected beyond this time frame (Wessmann et al., 2009). Seizures have been described in dogs with strokes but in general their frequency of occurrence seems low.
Twenty-seven dogs confirmed clinically to have experienced ischaemic stoke were evaluated for clinical topographic similarities with the human disease (Gredal et al., 2013). Seizures were reported as part of the acute symptomatology in 15 dogs (56%). Five of these were only reported with seizures in the acute phase, whereas seven developed chronic epilepsy after the stroke event. Four of five dogs (Paul et al., 2010), one of 16 dogs (Goncalves et al., 2011), one of 22 (Garosi et al., 2006) and zero of six dogs (Rossmeisl et al., 2007) with ischaemic stroke affecting the prosencephalon exhibited seizures at presentation; however, several dogs had seizure activity on follow-up examination. In humans, seizures have been reported to be the initial manifestation of stroke in 4–43% of patients depending on whether the cause is ischaemic or haemorrhagic (Giroud et al., 1994). Most humans that develop seizures following acute ischaemic events do so within 1 month of diagnosis.
reactive Seizures
Apart from epilepsy, seizures may represent a natural response of the normal brain to metabolic disturbances or intoxication (Engel and Starkman, 1994; Podell et al., 1995; Brauer et al., 2011). A study of 608 dogs with recurrent epileptic seizures documented 196 dogs (32%), which exhibited such ‘reactive’ seizures, compared to 154 dogs (25%) with structural epilepsy and 258 dogs (42%) with idiopathic/cryptogenic epilepsy (Fig. 7.1) (Steinmetz et al., 2013). An earlier study identified 29 dogs with reactive seizures
(a) Proportion of cases of epilepsy in dogs by aetiology
Dual pathology Cortical dysgenesis
(1.5%)
Others
Brain infections (6.1%) Brain tumours (8%)
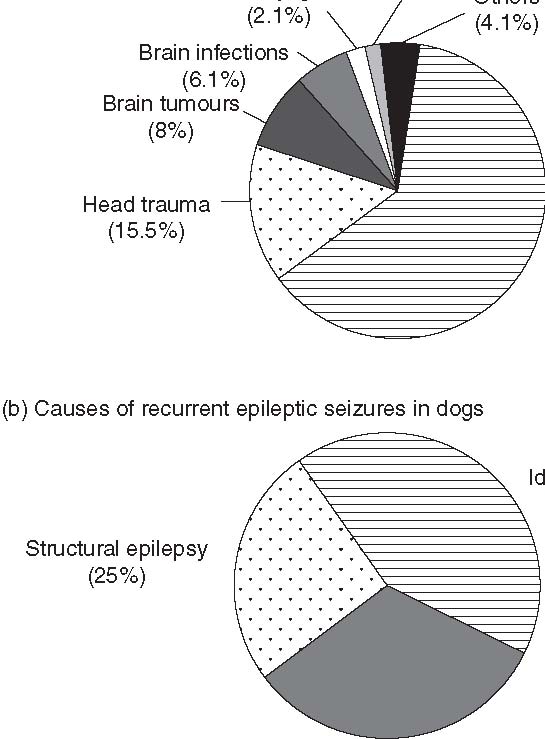 Idiopathic/cryptogenic
Idiopathic/cryptogenic
Head trauma (63%)
(15.5%)
Idiopathic/cryptogenic epilepsy (42%)
Structural epilepsy (25%)
Reactive (metabolic/toxic) (32%)
Fig. 7.1. Presumed causes of recurrent epileptic seizures in dogs. (a) Causes of seizures are illustrated for the 412 dogs with epilepsy. (b) The proportion of dogs (n = 196) with ‘reactive’ (provoked) seizures due to metabolic disturbances or intoxication shown in comparison to the proportions of dogs with structural (n = 154) or idiopathic/cryptogenic (n = 258) epilepsy within the whole group of 608 dogs with epileptic seizures (Steinmetz et al., 2013). Figure reproduced from Platt and Garosi (2012) with permission from Taylor & Francis Group, Abingdon.
out of a group of 240 (12%) with recurrent seizures (Pakozdy et al., 2008).
A recent study evaluated a total of 877 dogs, which had seizure disorders and were presented to a university hospital in Europe over a 4-year period (Brauer et al., 2011). A metabolic or toxic disorder (Fig. 7.2) was found in 96/877 (11%) of dogs with seizures. The majority of the 96 dogs had generalized seizures with loss of consciousness (49%, 47/96). Thirty-nine dogs (41%) were presented in status epilepticus. Thirty-one dogs (32%) were found to be hypoglycaemic.
Electrolyte disorders were responsible for seizures in ten dogs (10%). Hepatic encephalopathy with concurrent seizures occurred in nine dogs (9%). Hypothyroidism was the suspected cause in three dogs (3%). Uraemic encephalopathy (2%), presumptive hypoxia (2%) and hyperglycaemia (2%) were less frequent causes of seizures. Intoxication was the most frequent diagnosis (39%, 37/96). Metaldehyde (19%, 7/37) and organophosphate or carbamate poisoning (16%, 6/37) were the most frequent intoxications (Brauer et al., 2011).
Hypoglycaemia Electrolyte disorder
31 (32.3%) 10 (10.4%) Hepatic encephalopathy 9 (9.4%)
Hypothyroidism
3 (3.1%) Uraemic encephalopathy 2 (2.1%)
Hypoxia
2 (2.1%)
Hyperglycaemia37 (38.5%)
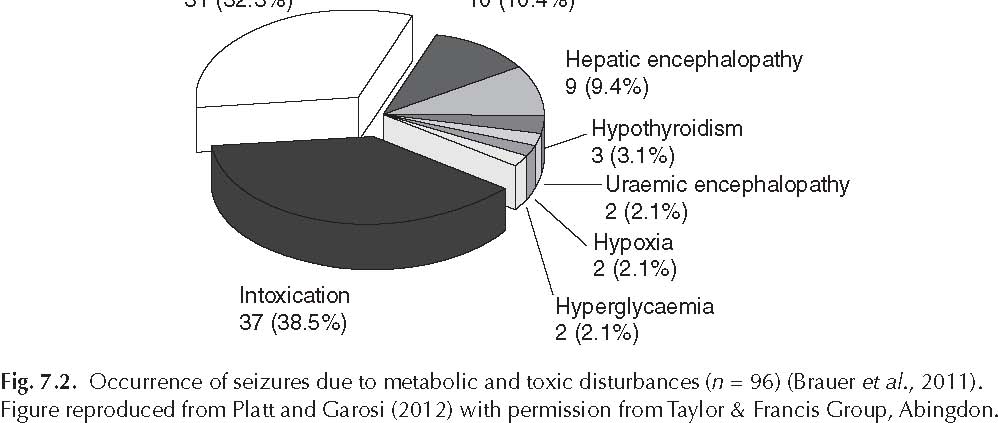
2 (2.1%)
Signalment and Canine Seizure Disorders
Breed
Based on pedigree analysis, a genetic basis for IE is suspected in a number of breeds, including the beagle, Belgian tervueren, keeshond, dachshund, German shepherd, Labrador retriever, golden retriever, Shetland sheepdog, Irish wolfhound, vizsla, Bernese mountain dog, horaks laborhounds, cocker spaniels, wirehaired fox terriers, boxers, dachshunds, toy poodles, Irish setters, miniature schnauzers, Siberian huskys, St Bernards, Bernese mountain dogs and English springer spaniels (Bielfelt et al., 1971; Hall and Wallace, 1996; Srenk and Jaggy, 1996; Famula et al., 1997; Jaggy et al., 1998; Kathmann et al., 1999; Thomas, 2000; Morita et al., 2002; Patterson et al., 2003, 2005; Casal et al., 2006).
A UK-based study was recently reported which evaluated the epidemiology of IE (Short et al., 2011). The diagnosis of IE was based on three or more generalized/focal or myoclonic seizures more than 24 h apart. Dogs were excluded from the study if they suffered from acquired epilepsy or reactive epilepsy, if they had had fewer than three seizures in total, and if less than 12 months had passed since the start of seizures and a brain MRI had not been performed; 1260 dogs satisfied the inclusion criteria, which comprised 79 pedigree breeds and a group of crossbreeds. Four breeds and cross-breed dogs combined to represent more than 50% of the total cases; the Labrador retriever (11%), Border collie (10.5%), Staffordshire bull terrier (5.2%) and the cross-breeds (20.5%). The study did recognize that the Labrador and German shepherd dog are popular breeds in the UK, with an excess of 40,000 and 20,000 dogs being registered with the UK Kennel Club, respectively, on a yearly basis; hence, a larger proportion of these breeds would be expected to have epilepsy due to their overall popularity (Short et al., 2011). The Border collie and Staffordshire bull terrier, however, have lower annual registration figures, with 3000 and 10,000 dogs, respectively, registered annually. The proportion of epileptic dogs in this series was similar for the Labrador (11%) and the Border collie (10.5%), and for the German shepherd dog (6.5%) and the Staffordshire bull terrier (5.2%), despite the difference in annual registration figures, suggesting that the Border collie and Staffordshire bull terrier are at a greater risk of developing epilepsy (Short et al., 2011).
Another recent UK-based epidemiological study evaluated dogs with epilepsy of unknown origin (EUO), which was diagnosed based on a history of more than two seizures in the absence of other medical problems and a history of seizures for more than 1 year, or four or more separate repeat anti-epileptic drug prescriptions (in the absence of evidence of their use for any purpose other than seizure control) (Kearsley-Fleet et al., 2013).
The study included 87,317 dogs presenting to veterinarians in the UK over a 14-month time period. Twenty per cent were crossbreeds. The most common pure dog breeds were the Labrador retriever (9.8%), Staffordshire bull terrier (7.8%), Parsons (Jack) Russell terrier (7.1%) and cocker spaniel (4.0%). The most common breed groups were the terrier group (22.2%), gundogs (21.4%), toy group (10.7%) and pastoral (8.5%). Of purebreds, 20.2% and 27.4% were small and large-sized, respectively, and 44.2% and 7.4% were short- and long-haired. Many (42.6%) of the dogs were solid coat-colour; 11.6% were black and 11.4% were red-golden, while 37.4% of dogs were two-tone in colour and 19.9% were multicoloured (Kearsley-Fleet et al., 2013).
Individual breeds also appeared at increased odds of EUO, though purebred dogs (as a group) versus crossbred dogs were not statistically significant in the univariable analysis. Heavier dogs were statistically significantly associated with presence of EUO, as were short coat length and certain coat colours in purebred dogs. In the multivariable analysis, sex, breed and age at consultation were statistically significantly associated with presence of EUO. Among specific breeds, the border terrier had 2.70 and the German shepherd dog 1.90 times the odds of EUO compared with crossbred dogs. The West Highland white terrier had reduced odds of 0.23 of EUO compared with crossbred dogs (Kearsley-Fleet et al., 2013).
Gender
The study by Kearsley-Fleet et al. (2013) found that in EUO, 48% of all dogs were female and 53.0% were neutered. Univariate risk factor analysis identified that males and neutered dogs appeared at increased odds of EUO. Male dogs had 1.72 times the odds of EUO compared with females. However, there was no evidence of an association between neuter status and EUO.
Short et al. (2011) investigated the gender and neuter status of a cohort of idiopathic epileptic dogs (n = 1188) and compared these data to randomly chosen non-epileptic dogs requiring veterinary attention (n = 2040).
The epileptic group contained 63% male dogs, compared with 51% of the non-epileptic population (P = 0.001), and 57% of the epileptic dogs were neutered, compared with 45% of the non-epileptic population (P = 0.001). When considering sex and neuter status, 49% of the male epileptic cases were neutered compared with 30% of the non-epileptic dogs (P = 0.001) and 69% of the female epileptic cases were neutered compared with 61% of the non-epileptic dogs (P = 0.01). The over-representation of males in the epileptic cohort is in agreement with other studies conducted on the Bernese mountain dog and the Irish wolfhound, although the latter study only showed a male bias before the age of 30 months (Casal et al., 2006; Kathmann et al., 1999). The age of neutering and the age of onset of epilepsy were not available for the dogs in the Short et al. study. A study of 49 idiopathic epileptic Border collies revealed that affected dogs were equally distributed between males (24 males; 12 neutered) and females (25 females; 18 neutered) (Hulsmeyer et al., 2010). Of 15 dogs neutered after seizure onset, only one owner (7%; 1/15) reported mild improvement in seizure frequency, whereas 13 dogs (86%; 13/15) exhibited no positive trends, and in one dog (7%; 1/15) the seizure frequency increased. Four dogs (neutered before seizure onset) experienced their first seizure at the time of neutering (two dogs experienced seizures on the same day and two dogs did so within 1 week after neutering) (Hulsmeyer et al., 2010). A study of Danish Labrador retrievers also found there to be no evidence for a gender predisposition, nor could any effect of neutering be identified (Berendt et al., 2002).
Age
Short et al. (2011) investigated the age of a cohort of idiopathic epileptic dogs (n = 1188) and compared these data to randomly chosen non-epileptic dogs requiring veterinary attention (n = 2040) and the distributions were similar; however, the age of onset of the seizure activity was not available (Short et al., 2011). The Kearsley-Fleet et al. (2013) study found that in EUO, dogs aged between 3.01 years and 6.00 years had 0.58 times the odds of EUO compared with dogs aged 10.01 years or older (Kearsley-Fleet et al., 2013).
In a study of epilepsy in dogs less than 1 year of age, the mean age at first seizure for the idiopathic group was 6.8 months (median seven), for the symptomatic group 7.5 months (median 7.5), the reactive group 4 months (median 3.25) and the probable symptomatic group (cryptogenic) 3.3 months (median 3.5) (Arrol et al., 2012). Overall, there was no association between seizure aetiology and age at which the first seizure occurred (Arrol et al., 2012).
Multiple studies have evaluated age of onset of IE in specific breeds. Jaggy et al. (1998) investigated IE in Labrador retrievers, finding that the mean age at first seizure of all epileptic dogs was 30.6 months with 31.9 months for males and 28.8 months for females (Jaggy et al., 1998). In contrast, mean age at first seizure of affected dogs from two healthy parental animals and from an epileptic and healthy parental animal was 35.1 and 22.1 months, respectively. Forty-one per cent of animals manifested the first seizure between 1 and 3 years of age, 25% within the first year and 34% at over 3 years (Jaggy et al., 1998). Labrador retrievers were also investigated as part of a separate epidemiological study; the age at the first observed seizure ranged between 5 and 91 months (Heynold et al., 1997). Approximately two-thirds of the dogs were between 1 and 3 years old, with an average of 34 months for males and 28 months for females (Heynold et al., 1997). At first presentation, the dogs were between 10 and 125 months of age (mean, 46). In a study of 49 Border collies with IE, age at seizure onset was between 1 and 5 years in 36 dogs (74%), <1 year in nine dogs (18%) and >5 years of age in four dogs (8%) (Hulsmeyer et al., 2010). Median age at seizure onset was 2.37 years regardless of gender and reproductive status. A large study involving 146 Irish wolfhounds with IE found that the incidence of females with onset of seizures between 3 and 4 years of age was low, with 82% of first observed seizures occurring before 3 years of age (Casal et al., 2006). The age of onset for males ranged from 6 to 107 months. Only 67% of males experienced their first seizure before the age of 3 years, whereas the onset of seizures occurred by 4 years of age in 83% of all male dogs (Casal et al., 2006). Overall, significantly more dogs had their first seizure before 3 years of life than after 3 years. Significantly more males than females had their first seizure in the 37- to 42-month and the 43- to 48-month periods (Casal et al., 2006).
Neurological Examination and Canine Seizure Disorders
A normal neurological examination in the interictal period is one of the criteria used to identify IE. However, while the majority of canine IE patients are completely normal between seizures and do not display any other clinical signs, others may express mild abnormalities, such as episodic ataxia, between seizures (Jokinen et al., 2007). Likewise, human IE patients may also display such symptoms between seizures (Imbrici et al., 2004). In human medicine, an increasing number of studies have identified systemic and neurobehavioural or psychiatric illnesses associated with epilepsy (Gaitatzis et al., 2004; Tellez-Zenteno et al., 2005; Nuyen et al., 2006; Austin and Caplan, 2007; LaFrance et al., 2008). In fact in people with a history of major depression or anxiety there is an increased risk of unprovoked seizures and epilepsy confirming a bidirectional relationship (Heinrichs and Seyfried, 2006; Kanner, 2006). Neurobehavioural co-morbidities have also been reported and studied in a variety of rodent models of epilepsy (Heinrichs and Seyfried, 2006). In a recent study, neurobehavioural changes were found to be related not only to epilepsy but also to pharmacological response, with pharmacoresistant rats having greater behaviour changes (Gastens et al., 2008).
A recent veterinary study investigated dogs with IE for associated behavioural changes (Shihab et al., 2011). The aim of the study was to look for behavioural changes associated with the development of epilepsy in dogs. Owners of a dog diagnosed with IE (n = 80) completed a behavioural and seizure questionnaire. Drug-naïve dogs showed an increase in the behaviour factors: (i) Fear/Anxiety;
(ii) Defensive Aggression; and (iii) Abnormal Perception. In dogs receiving anti-epileptic medication (AEM), there were still increases in Fear/Anxiety and Abnormal Perception, but no longer in Defensive Aggression.Additionally, increases in the following were also observed:
(i) Abnormal Reactivity; (ii) Attachment Disorder;
(iii) Demented Behaviour; and (iv) Apathetic Behaviour. Pharmacoresistant dogs had larger increases in Controlling Aggression, Abnormal Perception and Demented Behaviour than those dogs which were considered to be drug responders (Shihab et al., 2011).
A study conducted by Smith et al. (2008) investigated how many dogs with seizures and a normal neurological examination had abnormal MRI findings (Smith et al., 2008). This study found that in dogs under 6 years of age, an underlying cause was identified on MRI in only 2.2% of dogs, whereas this number increased to 26.7% of dogs over the age of 6 years. The study looking at dogs whose first seizure occurred below the age of 1 year demonstrated that 5.3% perceived to be clinically normal by neurological examination and blood results had an underlying cause identified with MRI and CSF analyses (Arrol et al., 2012). This result should help guide general practitioners when considering whether referral is indicated, especially in cases where financial constraints may be an issue.
In a study on dogs with epilepsy, which were less than 1 year of age, 74% (17 of 23) of the symptomatic dogs were found to have an abnormal neurological examination (Arrol et al., 2012). The odds ratio for dogs with a symptomatic aetiology having an abnormal neurological examination compared with the other groups combined was 76.5 (P = 0.001). Six dogs with a normal neurological examination had an underlying cause identified with MRI and CSF analyses.
Prognosis of Canine Seizure Disorders
Previous studies have shown that the lifespan of dogs with epilepsy have an increased risk of premature death compared with the general dog population (Proschowsky et al., 2003; Berendt et al., 2007; Hulsmeyer et al., 2010). This also applies for people with epilepsy for whom the risk of death is highest shortly after onset of seizures (Hauser et al., 1980; Neligan et al., 2011). The median survival time after index seizure is 2.07 years in Border collies and
2.3 years in a population of different purebred and mixed-breed dogs (Proschowsky et al., 2003, Berendt et al., 2007; Hulsmeyer et al., 2010). The life expectancy for Irish wolfhounds is shortened by almost 2 years in epileptic dogs compared with seizure-free relatives. The same study reported that more than 60% of the total number of deaths in the population were related to epilepsy (Casal et al., 2006).
The survival analysis in a study of epilepsy in juvenile dogs (<1 year old) gave a mean survival time of 7.1 years, which is substantially longer than that previously mentioned (Arrol et al., 2012). From these data the authors speculated that dogs with a younger age of seizure onset are easier to control with AEDs or have a less severe epilepsy course and thus are less likely to be euthanized. The IE group in this study had a similar outcome with a mean survival time of 6.1 years.
Factors such as gender, seizure onset, seizure frequency and seizure control might influence the lifespan of dogs with epilepsy. In the study of dogs with epilepsy which were less than 1 year of age, univariate analysis revealed that there was no significant association between age at first seizure and survival (P = 0.25), nor was there an association among sex (P = 0.50), neutered status (P = 0.40), breed (P = 0.27), whether the animal had experienced status epilepticus (P = 0.50), or presented primarily with focal (P = 0.17) or generalized seizures (P = 0.117) (Arrol et al., 2012). There was, however, a significant association between aetiology and survival (P = 0.01). The mean survival of dogs with idiopathic disease was 2255 days, compared to those with symptomatic disease which was 1855 days and reactive seizures which was 2744 days (Fig 7.2). There was also an association between cluster seizures and survival; the mean survival of those with cluster seizures was 2372 days, while those without cluster seizures had a median of 3061 days (P = 0.022). Finally, the number of AEMs the animal was receiving before investigation was also found to be associated with survival (P = 0.002). Dogs that had received no AEMs before investigation had a mean survival of 3287 days whereas those medicated with one AEM had a mean survival of 1575 days and those with two or more AEMs had a mean survival of 2169 days.
In another study, bitches lived longer with epilepsy compared with males, and daily treatment with anti-epileptic drugs did not influence the lifespan of dogs with epilepsy compared with dogs with epilepsy left untreated (Proschowsky et al., 2003). In Border collies, seizure onset before the age of 2 years has been shown to significantly decrease survival time (Hulsmeyer et al., 2010). A recent longitudinal study of survival in Belgian shepherds with IE revealed that the lifespan of epileptic dogs was not significantly shortened by the presence of epilepsy (Gullov et al., 2012). However, epilepsy was the predominant cause of death in the population (19/75 = 25%) and epilepsy-related deaths accounted for 70% (19/27) of all deaths in the group of dogs with epilepsy (Gullov et al., 2012). Two probable sudden unexpected deaths related to epilepsy occurred in dogs with generalized seizures.
Sudden unexpected death in epilepsy (SUDEP) is the most common cause of death directly related to epilepsy in humans, with an increased frequency of unexpected deaths up to 24 times compared with unexpected deaths in non-epileptics (Ficker et al., 1998; Surges et al., 2009). The mechanisms of SUDEP remain elusive, and it seems unlikely that the identification of a single mechanism will explain all incidents, as SUDEP is more likely a result of several predisposing and triggering factors (Surges et al., 2009; Donner, 2011). Poorly controlled chronic epilepsy with tonicclonic seizures seems to be the most well-defined risk factor for SUDEP in humans (Surges et al., 2009; Donner, 2011).
references
Agrawal, A., Timothy, J., Pandit, L. and Manju, M. (2006) Post-traumatic epilepsy: an overview. Clinical Neurology and Neurosurgery 108, 433–439.
Arrol, L., Penderis, J., Garosi, L., Cripps, P., Gutierrez-Quintana, R. and Goncalves, R. (2012) Aetiology and long-term outcome of juvenile epilepsy in 136 dogs. Veterinary Record 170, 335.
Austin, J.K. and Caplan, R. (2007) Behavioral and psychiatric comorbidities in pediatric epilepsy: toward an integrative model. Epilepsia 48, 1639–1651.
Bagley, R.S. and Gavin, P.R. (1998) Seizures as a complication of brain tumors in dogs. Clinical Techniques in Small Animal Practice 13, 179–184.
Bagley, R.S., Gavin, P.R., Moore, M.P., Silver, G.M., Harrington, M.L. and Connors, R.L. (1999) Clinical signs associated with brain tumors in dogs: 97 cases (1992-1997). Journal of the American Veterinary Medical Association 215, 818–819.
Beaumont, A. and Whittle, I.R. (2000) The pathogenesis of tumour associated epilepsy. Acta Neurochir (Wien) 142, 1–15.
Bellumori, T.P., Famula, T.R., Bannasch, D.L., Belanger, J.M. and Oberbauer, A.M. (2013) Prevalence of inherited disorders among mixed-breed and purebred dogs: 27,254 cases (1995-2010). Journal of the American Veterinary Medicine Association 242, 1549–1555.
Berendt, M. and Gram, L. (1999) Epilepsy and seizure classification in 63 dogs: a reappraisal of veterinary epilepsy terminology. Journal of Veterinary Internal Medicine 13, 14–20.
Berendt, M., Gredal, H., Pedersen, L.G., Alban, L. and Alving, J. (2002) A cross-sectional study of epilepsy in Danish Labrador Retrievers: prevalence and selected risk factors. Journal of Veterinary Internal Medicine 16, 262–268.
Berendt, M., Gredal, H. and Alving, J. (2004) Characteristics and phenomenology of epileptic partial seizures in dogs: similarities with human seizure semiology. Epilepsy Research 61, 167–173.
Berendt, M., Gredal, H., Ersboll, A.K. and Alving, J. (2007) Premature death, risk factors, and life patterns in dogs with epilepsy. Journal of Veterinary Internal Medicine 21, 754–759.
Berendt, M., Gullov, C.H., Christensen, S.L., Gudmundsdottir, H., Gredal, H., Fredholm, M. and Alban, L. (2008) Prevalence and characteristics of epilepsy in the Belgian shepherd variants Groenendael and Tervueren born in Denmark 1995-2004. Acta Veterinaria Scandinavica 50, 51.
Berendt, M., Gullov, C.H. and Fredholm, M. (2009) Focal epilepsy in the Belgian shepherd: evidence for simple Mendelian inheritance. Journal of Small Animal Practice 50, 655–661.
Bielfelt, S.W., Redman, H.C. and Mcclellan, R.O. (1971) Sire- and sex-related differences in rates of epileptiform seizures in a purebred beagle dog colony. American Journal of Veterinary Research 32, 2039–2048.
Brauer, C., Jambroszyk, M. and Tipold, A. (2011) Metabolic and toxic causes of canine seizure disorders: A retrospective study of 96 cases. Veterinary Journal 187, 272–275.
Casal, M.L., Munuve, R.M., Janis, M.A., Werner, P. and Henthorn, P.S. (2006) Epilepsy in Irish Wolfhounds. Journal of Veterinary Internal Medicine 20, 131–135.
Cornwell, H.J., Thompson, H., Mccandlish, I.A., Macartney, L. and Nash, A.S. (1988) Encephalitis in dogs associated with a batch of canine distemper (Rockborn) vaccine. Veterinary Record 122, 54–59.
D’Ambrosio, R. and Perucca, E. (2004) Epilepsy after head injury. Current Opinion in Neurology 17, 731–735.
Donner, E.J. (2011) Explaining the unexplained; expecting the unexpected: where are we with sudden unexpected death in epilepsy? Epilepsy Currents 11, 45–49.
Engel, J., Jr, and Starkman, S. (1994) Overview of seizures. Emerging Medicine Clinics of North America 12, 895–923.
Famula, T.R., Oberbauer, A.M. and Brown, K.N. (1997) Heritability of epileptic seizures in the Belgian tervueren. Journal of Small Animal Practice 38, 349–352.
Ficker, D.M., So, E.L., Shen, W.K., Annegers, J.F., O’Brien, P.C., Cascino, G.D. and Belau, P.G. (1998) Population-based study of the incidence of sudden unexplained death in epilepsy. Neurology 51, 1270–1274.
Fluehmann, G., Doherr, M.G. and Jaggy, A. (2006) Canine neurological diseases in a referral hospital population between 1989 and 2000 in Switzerland. Journal of Small Animal Practice 47, 582–587.
Friedenberg, S.G., Butler, A.L., Wei, L., Moore, S.A. and Cooper, E.S. (2012) Seizures following head trauma in dogs: 259 cases (1999-2009). Journal of the American Veterinary Medicine Association 241, 1479–1483.
Gaitatzis, A., Carroll, K., Majeed, A. and W. Sander, J. (2004) The epidemiology of the comorbidity of epilepsy in the general population. Epilepsia 45, 1613–1622.
Garosi, L., Mcconnell, J.E., Platt, S.R., Barone, G., Baron, J.C., De Lahunta, A. and Schatzberg, S.J. (2005) Results of diagnostic investigations and long-term outcome of 33 dogs with brain infarction (2000-2004). Journal of Veterinary Internal Medicine 19, 725–731.
Garosi, L., McConnell, J.F., Platt, S.R., Barone, G., Baron, J.C., De Lahunta, A. and Schatzberg, S.J. (2006) Clinical and topographic magnetic resonance characteristics of suspected brain infarction in 40 dogs. Journal of Veterinary Internal Medicine 20, 311–321.
Garosi, L.S. (2010) Cerebrovascular disease in dogs and cats. Veterinary Clinics of North America Small Animal Practice 40, 65–79.
Garosi, L.S. and McConnell, J.F. (2005) Ischaemic stroke in dogs and humans: a comparative review. Journal of Small Animal Practice 46, 521–529.
Gastens, A.M., Brandt, C., Bankstahl, J.P. and Loscher, W. (2008) Predictors of pharmacoresistant epilepsy: pharmacoresistant rats differ from pharmacoresponsive rats in behavioral and cognitive abnormalities associated with experimentally induced epilepsy. Epilepsia 49, 1759–1776.
Giroud, M., Gras, P., Fayolle, H., Andre, N., Soichot, P. and Dumas, R. (1994) Early seizures after acute stroke: a study of 1,640 cases. Epilepsia 35, 959–964.
Goncalves, R., Carrera, I., Garosi, L., Smith, P.M., Fraser Mcconnell, J. and Penderis, J. (2011) Clinical and topographic magnetic resonance imaging characteristics of suspected thalamic infarcts in 16 dogs. Veterinary Journal 188, 39–43.
Gredal, H., Skerritt, G.C., Gideon, P., Arlien-Soeborg, P. and Berendt, M. (2013) Spontaneous ischaemic stroke in dogs: clinical topographic similarities to humans. Acta Neurologia Scandanavica.
Gullov, C.H., Toft, N., Baadsager, M.M. and Berendt, M. (2011) Epilepsy in the Petit Basset Griffon Vendeen: prevalence, semiology, and clinical phenotype. Journal of Veterinary Internal Medicine 25, 1372–1378.
Gullov, C.H., Toft, N. and Berendt, M. (2012) A longitudinal study of survival in Belgian Shepherds with genetic epilepsy. Journal of Veterinary Internal Medicine 26, 1115–1120.
Hall, S.J. and Wallace, M.E. (1996) Canine epilepsy: a genetic counselling programme for keeshonds. Veterinary Record 138, 358–360.
Hauser, W.A., Annegers, J.F. and Elveback, L.R. (1980) Mortality in patients with epilepsy. Epilepsia 21, 399–412.
Heinrichs, S.C. and Seyfried, T.N. (2006) Behavioral seizure correlates in animal models of epilepsy: a road map for assay selection, data interpretation, and the search for causal mechanisms. Epilepsy and Behavior 8, 5–38.
Heynold, Y., Faissler, D., Steffen, F. and Jaggy, A. (1997) Clinical, epidemiological and treatment results of idiopathic epilepsy in 54 labrador retrievers: a long-term study. Journal of Small Animal Practice 38, 7–14.
Holliday, T.A., Cunningham, J.G. and Gutnick, M.J. (1970) Comparative clinical and electroencephalographic studies of canine epilepsy. Epilepsia 11, 281–292.
Hulsmeyer, V., Zimmermann, R., Brauer, C., Sauter-Louis, C. and Fischer, A. (2010) Epilepsy in Border Collies: clinical manifestation, outcome, and mode of inheritance. Journal of Veterinary Internal Medicine 24, 171–178.
Imbrici, P., Jaffe, S.L., Eunson, L.H., Davies, N.P., Herd, C., Robertson, R., Kullmann, D.M. and Hanna, M.G. (2004) Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain 127, 2682–2692.
Jaggy, A. and Bernardini, M. (1998) Idiopathic epilepsy in 125 dogs: a long-term study. Clinical and electroencephalographic findings. Journal of Small Animal Practice 39, 23–29.
Jaggy, A., Faissler, D., Gaillard, C., Srenk, P. and Graber, H. (1998) Genetic aspects of idiopathic epilepsy in Labrador retrievers. Journal of Small Animal Practice 39, 275–280.
Jokinen, T.S., Metsahonkala, L., Bergamasco, L., Viitmaa, R., Syrja, P., Lohi, H., Snellman, M., Jeserevics, J. and Cizinauskas, S. (2007) Benign familial juvenile epilepsy in Lagotto Romagnolo dogs. Journal of Veterinary Internal Medicine 21, 464–471.
Kanner, A.M. (2006) Depression and epilepsy: a new perspective on two closely related disorders. Epilepsy Currents 6, 141–146.
Kathmann, I., Jaggy, A., Busato, A., Bartschi, M. and Gaillard, C. (1999) Clinical and genetic investigations of idiopathic epilepsy in the Bernese mountain dog. Journal of Small Animal Practice 40, 319–325.
Kearsley-Fleet, L., O’Neill, D.G., Volk, H.A., Church, D.B. and Brodbelt, D.C. (2013) Prevalence and risk factors for canine epilepsy of unknown origin in the UK. Veterinary Record 172, 338.
LaFrance, W.C., Jr, Kanner, A.M. and Hermann, B. (2008) Psychiatric comorbidities in epilepsy. International Review of Neurobiology 83, 347–383.
Levine, J.M., Fosgate, G.T., Porter, B., Schatzberg, S.J. and Greer, K. (2008) Epidemiology of necrotizing meningoencephalitis in Pug dogs. Journal of Veterinary Internal Medicine 22, 961–968.
Licht, B.G., Licht, M.H., Harper, K.M., Lin, S., Curtin, J.J., Hyson, L.L. and Willard, K. (2002) Clinical presentations of naturally occurring canine seizures: similarities to human seizures. Epilepsy and Behavior 3, 460–470.
Licht, B.G., Lin, S., Luo, Y., Hyson, L.L., Licht, M.H., Harper, K.M., Sullivan, S.A., Fernandez, S.A. and Johnston, E.V. (2007) Clinical characteristics and mode of inheritance of familial focal seizures in Standard Poodles. Journal of the American Veterinary Medical Association 231, 1520–1528.
Lieu, A.S. and Howng, S.L. (2000) Intracranial meningiomas and epilepsy: incidence, prognosis and influencing factors. Epilepsy Research 38, 45–52.
Liigant, A., Haldre, S., Oun, A., Linnamagi, U., Saar, A., Asser, T. and Kaasik, A.E. (2001) Seizure disorders in patients with brain tumors. European Neurology 45, 46–51.
Lowenstein, D.H. (2009) Epilepsy after head injury: an overview. Epilepsia 50(Suppl. 2), 4–9.
Lynam, L.M., Lyons, M.K., Drazkowski, J.F., Sirven, J.I., Noe, K.H., Zimmerman, R.S. and Wilkens, J.A. (2007) Frequency of seizures in patients with newly diagnosed brain tumors: a retrospective review. Clinical Neurology and Neurosurgery 109, 634–638.
Mahaley, M.S., Jr, and Dudka, L. (1981) The role of anticonvulsant medications in the management of patients with anaplastic gliomas. Surgical Neurology 16, 399–401.
March, P.A. (1998) Seizures: classification, etiologies, and pathophysiology. Clinical Techniques in Small Animal Practice 13, 119–131.
Morita, T., Shimada, A., Takeuchi, T., Hikasa, Y., Sawada, M., Ohiwa, S., Takahashi, M., Kubo, N., Shibahara, T., Miyata, H. and Ohama, E. (2002) Cliniconeuropathologic findings of familial frontal lobe epilepsy in Shetland sheepdogs. Canadian Journal of Veterinary Research 66, 35–41.
Neligan, A., Bell, G.S., Johnson, A.L., Goodridge, D.M., Shorvon, S.D. and Sander, J.W. (2011) The long-term risk of premature mortality in people with epilepsy. Brain 134, 388–395.
Nuyen, J., Schellevis, F.G., Satariano, W.A., Spreeuwenberg, P.M., Birkner, M.D., Van Den Bos, G.A. and Groenewegen, P.P. (2006) Comorbidity was associated with neurologic and psychiatric diseases: a general practice-based controlled study. Journal of Clinical Epidemiology 59, 1274–1284.
Pákozdy, A., Leschnik, M., Tichy, A.G. and Thalhammer, J.G. (2008) Retrospective clinical comparison of idiopathic versus symptomatic epilepsy in 240 dogs with seizures. Acta Veterinaria Hungarica 56, 471–483.
Patterson, E.E., Mickelson, J.R., Da, Y., Roberts, M.C., Mcvey, A.S., O’Brien, D.P., Johnson, G.S. and Armstrong, P.J. (2003) Clinical characteristics and inheritance of idiopathic epilepsy in Vizslas. Journal of Veterinary Internal Medicine 17, 319–325.
Patterson, E.E., Armstrong, P.J., O’Brien, D.P., Roberts, M.C., Johnson, G.S. and Mickelson, J.R. (2005) Clinical description and mode of inheritance of idiopathic epilepsy in English springer spaniels. Journal of the American Veterinary Medical Association 226, 54–58.
Paul, A.E., Lenard, Z. and Mansfield, C.S. (2010) Computed tomography diagnosis of eight dogs with brain infarction. Australian Veterinary Journal 88, 374–380.
Pitkanen, A., Immonen, R.J., Grohn, O.H. and Kharatishvili, I. (2009) From traumatic brain injury to posttraumatic epilepsy: what animal models tell us about the process and treatment options. Epilepsia 50 (Suppl. 2), 21–29.
Platt, S.R. and Garosi, L. (2003) Canine cerebrovascular disease: do dogs have strokes? Journal of the American Animal Hospital Association 39, 337–342.
Platt, S.R. and Garosi, L. (2012) Small Animal Neurological Emergencies, Taylor & Francis Group, Abingdon, UK.
Platt, S.R. and Haag, M. (2002) Canine status epilepticus: a retrospective study of 50 cases. Journal of Small Animal Practice 43, 151–153.
Platt, S.R., McConnell, J.F., Garosi, L.S., Ladlow, J., De Stefani, A. and Shelton, G.D. (2006) Magnetic resonance imaging in the diagnosis of canine inflammatory myopathies in three dogs. Veterinary Radiology and Ultrasound 47, 532–537.
Podell, M., Fenner, W.R. and Powers, J.D. (1995) Seizure classification in dogs from a nonreferral-based population. Journal of the American Veterinary Medical Association 206, 1721–1728.
Proschowsky, H.F., Rugbjerg, H. and Ersboll, A.K. (2003) Mortality of purebred and mixed-breed dogs in Denmark. Preventive Veterinary Medicine 58, 63–74.
Rossmeisl, J.H., Jr, Rohleder, J.J., Pickett, J.P., Duncan, R. and Herring, I.P. (2007) Presumed and confirmed striatocapsular brain infarctions in six dogs. Veterinary Ophthalmology 10, 23–36.
Schaller, B. and Ruegg, S.J. (2003) Brain tumor and seizures: pathophysiology and its implications for treatment revisited. Epilepsia 44, 1223–1232.
Schwartz, M., Lamb, C.R., Brodbelt, D.C. and Volk, H.A. (2011) Canine intracranial neoplasia: clinical risk factors for development of epileptic seizures. Journal of Small Animal Practice 52, 632–637.
Schwartz, M., Munana, K.R. and Nettifee-Osborne, J. (2013) Assessment of the prevalence and clinical features of cryptogenic epilepsy in dogs: 45 cases (2003-2011). Journal of the American Veterinary Medical Association 242, 651–657.
Shamji, M.F., Fric-Shamji, E.C. and Benoit, B.G. (2009) Brain tumors and epilepsy: pathophysiology of peritumoral changes. Neurosurgical Review 32, 275–284; discussion 284–286.
Shihab, N., Bowen, J. and Volk, H.A. (2011) Behavioral changes in dogs associated with the development of idiopathic epilepsy. Epilepsy and Behavior 21, 160–167.
Short, A.D., Dunne, A., Lohi, H., Boulton, S., Carter, S.D., Timofte, D. and Ollier, W.E. (2011) Characteristics of epileptic episodes in UK dog breeds: an epidemiological approach. Veterinary Record 169, 48.
Smith, P.M., Talbot, C.E. and Jeffery, N.D. (2008) Findings on low-field cranial MR images in epileptic dogs that lack interictal neurological deficits. Veterinary Journal 176, 320–325.
Snyder, J.M., Shofer, F.S., Van Winkle, T.J. and Massicotte, C. (2006) Canine intracranial primary neoplasia: 173 cases (1986-2003). Journal of Veterinary Internal Medicine 20, 669–675.
Srenk, P. and Jaggy, A. (1996) Interictal electroencephalographic findings in a family of golden retrievers with idiopathic epilepsy. Journal of Small Animal Practice 37, 317–321.
Steinmetz, S., Tipold, A. and Loscher, W. (2013) Epilepsy after head injury in dogs: a natural model of post-traumatic epilepsy. Epilepsia 54, 580–588.
Surges, R., Thijs, R.D., Tan, H.L. and Sander, J.W. (2009) Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nature Reviews - Neurology 5, 492–504.
Tellez-Zenteno, J.F., Matijevic, S. and Wiebe, S. (2005) Somatic comorbidity of epilepsy in the general population in Canada. Epilepsia 46, 1955–1962.
Thomas, W.B. (2000) Idiopathic epilepsy in dogs. Veterinary Clinics of North America - Small Animal Practice 30, 183–206, vii.
Tipold, A. (1995) Diagnosis of inflammatory and infectious diseases of the central nervous system in dogs: a retrospective study. Journal of Veterinary Internal Medicine 9, 304–314.
Van Breemen, M.S., Wilms, E.B. and Vecht, C.J. (2007) Epilepsy in patients with brain tumours: epidemiology, mechanisms, and management. Lancet Neurology 6, 421–430.
Weissl, J., Hulsmeyer, V., Brauer, C., Tipold, A., Koskinen, L.L., Kyostila, K., Lohi, H., Sauter-Louis, C., Wolf, M. and Fischer, A. (2012) Disease progression and treatment response of idiopathic epilepsy in Australian Shepherd dogs. Journal of Veterinary Internal Medicine 26, 116–125.
Wessmann, A., Chandler, K. and Garosi, L. (2009) Ischaemic and haemorrhagic stroke in the dog. Veterinary Journal 180, 290–303.
8 Epidemiology of Feline Seizures
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Prevalence of Seizures in Cats
The prevalence of feline seizures has been reported as 2.1% (164/7809) of all cats evaluated at a veterinary teaching hospital during a 5- year period (see Schriefl et al., 2008; Chapter 3), as 3.3% (235/7243) of the feline hospital population during about 6 years at another institution (see Pakozdy et al., 2010; Chapter 3) and as 14% (177/1258) of all cats evaluated during a 15-year period at another veterinary teaching hospital (Cizinauskas et al., 2003). The prevalence of recurrent seizures in cats with structural brain diseases has been reported as 0.1% (when evaluated in 1978) and 0.5% (when evaluated in 1985) of the hospital population in another study (see Schwartz-Porsche and Kaiser, 1989; Chapter 3). The prevalence of recurrent seizures has been reported as 22.5% (36/160) (Troxel et al., 2003) and 23% (14/61) (Tomek et al., 2006) in cats with a histological diagnosis of intracranial neoplasia and as 25% (14/55) in cats with histologically confirmed feline infectious peritonitis (FIP) (Timmann et al., 2008).
Prevalence of Feline Seizures Based on Semiological classification
Veterinary classification of seizures based on clinical manifestations is described in
Chapter 3 (see Table 3.2). Generalized-onset seizures have been reported as the most common type of seizure in the majority of studies on feline seizures (Schwartz-Porsche and Kaiser, 1989; Quesnel et al., 1997; Cizinauskas et al., 2003; Barnes et al., 2004; Pakozdy et al., 2010, 2013). Generalized-onset seizures have also been reported as the most common type of seizure in two studies that investigated the prevalence of seizures in cats with intracranial neoplasia and FIP, respectively (Tomek et al., 2006; Timmann et al., 2008). Ictal phenomenology was observed and described by the cat owners in the vast majority of cases and therefore focal clinical manifestations at the onset of the ictus may have gone unnoticed. Only two studies reported higher prevalence of focal-onset seizures (52% and 67%, respectively) compared to generalized-onset seizures in cats with various seizure aetiologies (see Volk et al., 2007; Schriefl et al., 2008). Secondary generalization occurred in 65% and 90% of these cats, respectively (see Volk et al., 2007; Schriefl et al., 2008; Chapter 3). Reported prevalence of seizure types classified based on clinical manifestations is summarized in Table 8.1. The clinical manifestations of different seizure types in cats are described in Chapter 3.
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
Table 8.1. Prevalence of feline seizure types based on semiological classification.
| Schwartz- | ||||||
|---|---|---|---|---|---|---|
| Porsche | Quesnel | Cizinauskas | Barnes | Schriefl | Pakozdy | Pakozdy |
| and Kaiser, | et al., | et al., | et al., | et al., | et al., | et al., |
| 1989 | 1997 | 2003a | 2004 | 2008 | 2010 | 2012a |
Focal-onset seizures with or without secondary generalization
Generalized-onset tonic-clonic seizures with or without status epilepticus
Generalized clonic seizures
Focal and generalized onset seizures
Unclassified seizures
Total number of cats
10 (34%)c 11 (35%) 4 (24%) 47 (52%) 8 (6%) 6 (17%)
26 (60%) 14 (46%) 20 (61%) 13 (76%) 44 (48%) 63 (51%) 17 (47%)
| 8 (20%)b | ||||||
|---|---|---|---|---|---|---|
| 6 (20%) | 2 (4%) | 54 (43%) 13 (36%) | ||||
| 8 (20%) | ||||||
| 42 | 30 | 33 | 17 | 91 | 125 | 36 |
aData on cats with idiopathic or presumed idiopathic epilepsy bSome or all of these may have been complex focal seizures with or without secondary generalization cAll these ten cats had complex focal seizures (including paroxysms of abnormal behaviour)
Prevalence of Feline Seizures Based on Aetiological Classification
Veterinary classification of seizures based on aetiological classification is described in Chapter 3 (see Table 3.3). Structural epilepsy has been reported as the most common type of feline epilepsy in several studies (see Quesnel et al., 1997; Cizinauskas et al., 2003; Schriefl et al., 2008; Wahle et al., 2014; Chapter 3). However, other studies have reported a higher prevalence of idiopathic epilepsy than structural epilepsy in cats (Rusbridge, 2005; Schwartz-Porsche and Kaiser, 1989; Pakozdy et al., 2010; Chapter 3). In addition, the same prevalence for structural and cryptogenic epilepsy (defined as epilepsy without any extracranial or identifiable intracranial disease that is not suspected to be genetic in origin) has been reported by others (see Barnes et al., 2004; Chapter 3). Reported prevalence of seizures/epilepsies classified based on their underlying aetiology is summarized in Table 8.2.
Comparison of different studies on feline seizures should be done with awareness of the following limitations: differences in terminology and classification of seizures and epilepsies, differences in inclusion criteria, variations in type and size of the population under investigation, geographic variation in disease prevalence, different duration of disease at time of evaluation, differences in components of the diagnostic investigation protocol (particularly the criteria to diagnose idiopathic epilepsy) and differences in follow-up methods and duration.
Overall, the reported prevalence of idiopathic epilepsy has ranged from 0% (see Quesnel et al., 1997; Barnes et al., 2004) to 59% (Schwartz-Porsche and Kaiser, 1989). This discrepancy is mainly due to the different definitions of idiopathic epilepsy. While some authors (see Quesnel et al., 1997; Barnes et al., 2004) have defined idiopathic epilepsy as recurrent seizures without any underlying structural brain lesion or extracranial disease that are presumably genetic in origin, others have defined idiopathic epilepsy as recurrent seizures of unknown origin after exclusion of metabolic, toxic and structural brain disorders (Schwartz-Porsche and Kaiser, 1989; Rusbridge, 2005; Schriefl et al., 2008; Pakozdy et al., 2010; Chapter 3). This latter definition is similar to Barnes’ definition of cryptogenic epilepsy (Barnes et al., 2004; Chapter 3). In addition,
Table 8.2. Prevalence feline seizures/epilepsies based on their aetiological classification.
| Idiopathic or | ||||||
|---|---|---|---|---|---|---|
| presumed | Unknown | |||||
| Reactive | Structural | idiopathic | Cryptogenic | seizure | Total number | |
| seizures | epilepsy | epilepsy | epilepsy | aetiology | of cats | |
| Schwartz-Porsche, | 17 (41%) | 25 (59%) | 42 | |||
| 1989 | ||||||
| Quesnel et al., 1997 | 26 (87%) | 4 (13%) | 30 | |||
| Cizinauskas | 26 (17%) | 73 (48%) | 33 (21%) | 22 (14%) | 154 | |
| et al., 2003 | ||||||
| Barnes et al., 2004 | 3 (18%) | 7 (41%) | 7 (41%) | 17 | ||
| Rusbridge, 2005 | 4% | 42% | 54% | Not | ||
| available | ||||||
| Schriefl et al., | 20 (22%) | 45 (50%) | 23 (25%) | 91a | ||
| 2008 | ||||||
| Pakozdy et al., | 35 (28%) | 43 (34%) | 47 (38%) | 125 | ||
| 2010 | ||||||
| Wahle et al., 2014 | 25 (31%) | 38 (47%) | 18 (22%) | 81 | ||
aCardiac syncope was diagnosed in 3 (3%) cats
the diagnostic protocol to rule out metabolic, toxic and structural brain disorders varies among studies and several cats reported with idiopathic epilepsy have not undergone brain imaging or post-mortem examination.
The prevalence of idiopathic epilepsy in cats has been reported to be lower than in dogs in the few studies that have investigated prevalence of seizure/epilepsy aetiology at the same institution (Rusbridge, 2005; Pakozdy et al., 2010). One study reported a prevalence of idiopathic epilepsy of 38% in cats and 48% in dogs (Pakozdy et al., 2010), and another study reported a prevalence of idiopathic epilepsy of 54% in cats and 68% in dogs (Rusbridge, 2005).
Seizure type (based on semiological classification) does not seem to be predictive of underlying seizure aetiology (Barnes et al., 2004; Schriefl et al., 2008; Pakozdy et al., 2010). No statistically significant association was identified between various types of seizures (focal-onset with or without secondary generalization, or generalized-onset) and different seizure aetiologies (reactive, structural or idiopathic) (Schriefl et al., 2008; Pakozdy et al., 2010). Although the percentage of cats with generalized-onset tonicclonic seizures appeared higher among those with idiopathic epilepsy compared to cats with other seizure aetiologies, this difference was not statistically significant. Likewise, although the percentage of cats with status epilepticus appeared higher in those with reactive seizures (6/20, 30%) or structural (10/45, 22%) seizures than in those with idiopathic epilepsy (1/23, 4%), differences were not statistically significant (see Schriefl et al., 2008). In contrast, another study reported that status epilepticus was significantly more common in cats with structural epilepsy or reactive seizures (31/78, 40%) than in cats diagnosed with idiopathic epilepsy (9/47, 19%) (see Pakozdy et al., 2010). Prevalence of cluster seizures was similar between cats with structural epilepsy or reactive seizures (46/78, 59%) and cats with idiopathic epilepsy (25/47, 53%) (see Pakozdy et al., 2010).
Signalment and Feline Seizure Disorders
Breed
The domestic short hair is the most represented breed in studies on feline seizures and most likely the most common breed in the respective hospital population (Quesnel et al., 1997; Barnes et al., 2004; Schriefl et al., 2008). However this information is provided only in one study in which domestic short hairs represented 93.6% (117 of 125) of cats with seizures and 82.1% (5946 of 7243) of the feline hospital population, whereas Persian cats represented 1.6% (2/125) of seizuring cats and 7.4% (533/7243) of the feline hospital population (Pakozdy et al., 2010). Domestic short hairs are also over-represented among cats with idiopathic epilepsy (Cizinauskas et al., 2003; Pakozdy et al., 2012; Wahle et al., 2014) making identification of a possible familial or genetic basis for feline idiopathic epilepsy very difficult to impossible.
Gender
The majority studies on feline seizures report no apparent gender predisposition for seizures or epilepsy (see Quesnel et al., 1997; Barnes et al., 2004; Schriefl et al., 2008; Pakozdy et al., 2012). No statistically significant difference in gender was identified between cats with idiopathic and structural epilepsy or reactive seizures (see Pakozdy et al., 2010; Wahle et al., 2014).
Age
Age at the onset of seizures varies among studies and overall ranges from 1 week to 22 years. Reported ages at onset of seizures are summarized in Table 8.3.
The age at seizure onset is significantly different between cats with idiopathic epilepsy and cats with structural or reactive epilepsy (see Schriefl et al., 2008; Pakozdy et al., 2010). In one study,
Table 8.3. Age at onset of seizures reported in different studies on feline seizures.
Ages at seizure onset
| All cats | Cats with | Cats with | Cats with | Cats with |
| included | reactive | structural | idiopathic | cryptogenic |
| in the study | seizures | epilepsy | epilepsy | epilepsy |
Schwartz-6m to >8y NR <2y and >8y 6m to >8ya NR Porsche, 1989
Quesnel et al., 3m–13y NR NA NR NR 1997 Cizinauskas 4m–13y NA NA 4m–13yb NR et al., 2003 Barnes et al., 6m–18y 2.1–5.3y 0.5–14.4y NR 1.2–18y (mean, 2004 (mean, 7.1 y)c 10.7y)d Schriefl et al., 1w–19y (mean, 8.2y) (mean, 8.1y) 1–12y (mean, NR 2008 (median, 5y) 3.5y; median 3y) Pakozdy et al., 4m–20y 0.3–20y (mean, 0.3–20y 0.5–14y (mean, NR 2010 8.4y)e (mean, 8.4y)e 4.6y) Pakozdy et al., 6m–11y (mean, NR NR 6m–11y (mean, NR 2012f 4.8±3.5y) 4.8±3.5y) Wahle et al., 0.3–22y 0.4–22y (median 0.3–19y (median 0.4–14.4y (median NA 2014 7.4y) 7.8y) 3.8y)
w, week; m, month/s; y, year/s; NA, not available; NR, none of the cats in this study was reported with this type of seizure/ epilepsy a60% of these cats were 6 months to 3 years old b64% of these cats were 1 to 6 years old cMean age at onset of seizures was 3.4 years for the four cats with meningoencephalitis and 12.1 years for the cats with cerebral neoplasia dAll but one of the cats were >5 years old eReactive seizures and structural epilepsy were grouped together as secondary epilepsy in this study. fThis study includes only cats with idiopathic epilepsy
cats younger than 7 years of age were more likely to have idiopathic epilepsy and those older than 7 years of age were more likely to have structural or reactive seizures (Pakozdy et al., 2010). In another study, cats diagnosed with idiopathic epilepsy were significantly younger (mean, 3.5 years) when the first seizure occurred than cats with reactive seizures (mean, 8.2 years) or with structural epilepsy (mean, 8.1 years) (Schriefl et al., 2008). Age at seizure onset was not significantly different between cats with structural epilepsy and cats with reactive seizures (Schriefl et al., 2008). Age at seizure onset and duration of the seizure disorder prior to examination are not significantly different between cats with structural and cryptogenic epilepsy (Barnes et al., 2004). Among cats with structural epilepsy, cats with meningoencephalitis (mean age 3.4 years) are generally younger than cats with neoplasia (mean age 12.1 years) (Barnes et al., 2004). Among cats with intracranial neoplasia, cats with lymphoma (median age 5 years, mean age 5.6 years) are significantly younger than cats with meningiomas (median age 11 years, mean age 10.3 years) (Tomek et al., 2006).
Neurological Examination and Feline Seizure Disorders
Interictal neurological abnormalities are significantly more common in cats with structural epilepsy and reactive seizures (57/78, 73%) than in cats with idiopathic epilepsy (2/47, 4%) (Pakozdy et al., 2010). Although a normal neurological examination is one of the diagnostic criteria for idiopathic epilepsy, interictal neurological abnormalities may sometimes occur during the post-ictal phase, secondary to hypoxic or excitoxic cerebral injury following severe or prolonged seizures, or due to anti-epileptic medication (AEM) side effects (see Seizure and AEM-associated neurological deficits, Chapter 10). Neurologic examinations should be repeated over time on these cats. Reported prevalence of interictal neurological abnormalities in cats with symptomatic (structural) epilepsy ranges from 19/30 (63%) (Quesnel et al., 1997) to 7/7 (100%) (Barnes et al., 2004). Therefore, similarly to dogs, the neurological examination may be normal in some seizuring cats, despite the presence of structural brain disease, particularly with lesions affecting the olfactory bulb and frontal lobes. Common neurological deficits in order of decreasing frequency include abnormal postural reactions, bilateral or unilateral menace deficits, obtundation, ataxia, decreased facial sensation and compulsive circling (Quesnel et al., 1997; Tomek et al., 2006; Schriefl et al., 2008).
Aetiologies of Feline Seizure Disorders
Reported aetiologies for reactive seizures and structural epilepsy in cats examined at referral institutions vary among different studies (Tables 8.4 and 8.5). Commonly reported aetiologies for reactive seizures include hepatic encephalopathy, severe uraemia with end-stage renal disease, hypoglycaemia (generally secondary to insulin overdose), hypertension, polycythaemia vera, and intoxication with permethrin or inhibitors of acetylcholinesterase. The most commonly reported aetiologies of structural epilepsy include brain neoplasia (mainly meningiomas), meningoencephalitis (mainly of unknown cause) and necrosis of hippocampus and piriform lobe. Hippocampal and piriform lobe necrosis has been reported predominantly in Switzerland and Austria and although an exogenous toxic aetiology was initially proposed (Fatzer et al., 2000), this hypothesis was not supported by further studies (Cizinauskas et al., 2003; Brini et al., 2004; Schmied et al., 2008; Pakozdy et al., 2010; Pakozdy et al., 2011). There has been discussion whether the necrosis of hippocampus and piriform lobe constitutes the cause or the consequence of severe seizures (Vanhaesebrouck et al., 2012). Hippocampal neurons are sensitive to hypoxia, hyperglycaemia, hypoglycaemia and glutamate excitotoxicity, which can all occur with prolonged seizure activity. In humans, ongoing loss of hippocampal neurons and astrocyte remodelling leading to hippocampal
Table 8.4. Prevalence of reported aetiologies for reactive seizures in cats examined at referral institutions.
Barnes et al., 2004 Schriefl et al., 2008 Pakozdy et al., 2010
| Hepatic encephalopathy | 3 (100%) | 4 (21%) | 2 (6%) |
| Severe uraemia with end-stage | 1 (5%) | 3 (9%) | |
| renal disease | |||
| Hypoglycaemia attributable to | 3 (16%) | ||
| insulin overdose | |||
| Hypoglycaemia | 1 (3%) | ||
| Diabetic ketoacidosis | 2 (6%) | ||
| Hyperosmolality | 2 (6%) | ||
| Severe hyperthyroidism with | 2 (11%) | 3 (9%) | |
| severe systemic hypertension | |||
| Hypertension | 2 (6%) | ||
| Polycythaemia vera/erythrocytosis | 2 (11%) | 4 (12%) | |
| Severe anaemia (hypoxia) | 1 (5%) | 2 (6%) | |
| Severe lung oedema (hypoxia) | 2 (6%) | ||
| Narcosis-induced hypoxia | 1 (3%) | ||
| Other extracranial disorder | 1 (3%) | ||
| Hypocalcaemia | 1 (5%) | ||
| Severe idiopathic | 1 (5%) | ||
| hypertriglyceridaemia | |||
| Iatrogenic hypoadrenocorticism | 1 (5%) | ||
| Permethrin intoxication | 2 (11%) | 4 (12%) | |
| Inhibitors of acetylcholinesterase | 1 (5%) | 4 (12%) | |
| intoxication | |||
| Total number of cats with reactive | 3 | 19a | 33 |
| seizures |
aIntoxication was strongly suspected in one additional cat but the specific aetiology was not identified
sclerosis is associated with initial precipitating injuries (e.g. trauma, hypoxia, infection and prolonged initial seizure) early in life and perpetuated by ongoing seizure activity (see Schriefl et al., 2008; Chapter 3). Similarly, feline hippocampal and piriform lobe necrosis may result from more than one aetiology (Pakozdy et al., 2014). Most recently, hippocampal necrosis has been reported in association with antibodies against voltage-gated potassium channel complexes, principally leucine-rich glioma inactivated 1 resulting in focal seizures characterized by orofacial involvement (Pakozdy et al., 2013) (Plate 22). In a recent post mortem study, hippocampal sclerosis was identified in approximately one third of epileptic cats with various seizure aetiologies (Wagner, 2014).
Prevalence of underlying seizure aetiologies in the non-referral feline population may differ to those presented in Tables 8.4 and 8.5. In addition, disease prevalence is likely to be influenced by geographic variation.
Prognosis of Feline Seizure Disorders
Survival times in seizuring cats are significantly affected by seizure aetiology (Barnes et al., 2004; Schriefl et al., 2008; Table 8.6).
The 1-year survival rate for cats with reactive seizures is significantly longer than that for cats with structural epilepsy. Cats with idiopathic epilepsy have a significantly longer 1-year survival rate than cats with reactive or structural seizures (Schriefl et al., 2008; Wahle et al., 2014). Cats with cryptogenic epilepsy have significantly longer survival times (mean 1.9 years) than cats with structural epilepsy (<3 months) (Barnes et al., 2004). In addition, cats with status epilepticus (most commonly associated with reactive or structural seizures) have a significantly shorter survival time than cats that do not have status epilepticus (Schriefl et al., 2008).
Table 8.5. Prevalence of reported aetiologies for structural epilepsy in cats examined at referral institutions.
Quesnel Cizinauskas Barnes Schriefl Pakozdy et al., 1997 et al., 2003 et al., 2004 et al., 2008 et al., 2010
| Neoplasia | 2 (8%) | 13 (18%) | 3 (43%) | 17 (37%) | 15 (35%) |
| Meningioma | 2 (8%) | 8 (18%) | 9 (22%) | ||
| Oligodendroglioma | 1 (2%) | 3 (7%) | |||
| Astroglioma | 1 (2%) | ||||
| Astrocytoma | 1 (14%) | 1 (2%) | |||
| Spindle cell tumour | 1 (2%) | ||||
| Lymphoma | 1 (14%) | 1 (2%) | |||
| Pituitary adenocarcinoma | 1 (14%) | ||||
| Osteosarcoma of the | 1 (2%) | ||||
| cranial bone | |||||
| Metastatic tumours | 6 (13%) | ||||
| Infectious/inflammatory | 15 (57%) | 21 (29%) | 4 (57%) | 20 (44%) | 7 (17%) |
| FIP | 2 (30%) | 6 (13%) | 2 (5%) | ||
| Meningoencephalitis | 14a (53%) | 1 (14%) | 9 (20%) | 5 (12%) | |
| of unknown origin | |||||
| Toxoplasmosis | 5 (11%) | ||||
| Cryptococcus neoformans | 1 (14%) | ||||
| Cerebral abscess | 1 (4%) | ||||
| Necrosis of hippocampus | 39 (53%) | 2 (5%) | 14 (34%) | ||
| and piriform lobe | |||||
| Ischaemic encephalopathy | 6 (23%) | 1 (2%) | |||
| Hypertensive angiopathy | 3 (7%) | ||||
| and encephalopathy | |||||
| Polycythaemia vera with | 2 (8%) | ||||
| secondary brain vascular | |||||
| lesions | |||||
| Brain trauma | 1 (4%) | 3 (7%) | 1 (2%) | ||
| Brain degeneration | 4 (10%) | ||||
| of unknown origin | |||||
| Total number of cats with | 26b | 73 | 7 | 45 | 42 |
| structural epilepsy |
aThe meningoencephalitis was non-suppurative in all cats bA definitive diagnosis could not be reached in four cats
Table 8.6. Survival times for cats diagnosed with reactive, structural and idiopathic seizures (see Schriefl et al., 2008).
Reactive seizures Structural epilepsy Idiopathic epilepsy
Mean 68w 24w 128w Median 35w 3w 148w Range 1d–216w 1d–192w 2w–284w Probability of survival at 50% 16% 82%
1 year post-diagnosis
d, days; w, weeks
Information on efficacy of anti-epileptic treatment in cats is limited. Phenobarbital (PB) is the most commonly used AEM and seems efficacious in cats with idiopathic and structural epilepsy (Quesnel et al., 1997; Pakozdy et al., 2012). In a recent study all 30 cats treated with PB achieved a ³50% reduction in the number of seizures; 13 became seizure free (Finnerty et al., 2014). Another recent study including 36 idiopathic epileptic cats with a minimum follow up of 1 year reported seizure eradication in approximately 45% of cats, good (1–5 seizures/ year) to moderate (6–10 seizures/year) seizure control in approximately 25% of cats, and poor (>10 seizures/ year) seizure control in 30% of cats following PB treatment. Four of these 36 cats were also administered diazepam, gabapentin or levetiracetam (Pakozdy et al., 2012). Similar results (44% seizure free, 31% adequate seizure control, 25% inadequate seizure control) were reported in another study including 16 epileptic cats (Volk et al., 2007). In the larger study, seizure duration and severity (based on owner assessment) decreased in 72% and 69% of cats, respectively. Quality of life was considered good by the owners in 72% of cats. Early initiation of PB treatment was significantly associated with a more favourable outcome than delayed treatment. Cats that achieved seizure eradication for over 1 year remained seizure free for a period of years, unless PB dosage was reduced or discontinued. Seizures recurred in 6/8 (86%) cats in which PB was reduced or discontinued after 1 seizure-free year. Age at seizure onset, body weight and gender are not associated with outcome (number of seizures per year) (Pakozdy et al., 2012). In a more recent study 44% (8/18) of cats with idiopathic epilepsy experienced seizure remission. Median seizure remission time was
1.4 years (range, 1.0–5.4 years). Seizure remission was maintained with (n = 5) or without AEM (n = 3) (Wahle et al., 2014).
In one study including predominantly cats with structural epilepsy, treatment of the underlying seizure aetiology as well as anti-epileptic treatment (PB and/or diazepam) resulted in seizure eradication or low seizure frequency in 57% (17/30) of cats with a follow up of 3 to 21 months (Quesnel et al., 1997). Seizure severity at presentation does not seem to be predictive of long-term response to anti-epileptic treatment in cats (Quesnel et al., 1997; Rusbridge, 2005). However, an aggressive diagnostic and therapeutic approach may improve prognosis and may be associated with favourable outcome (Wahle et al., 2014).
References
Barnes, H.L., Chrisman, C.L., Mariani, C.L., Sims, M. and Alleman, A.R. (2004) Clinical signs, underlying cause, and outcome in cats with seizures: 17 cases (1997-2002). Journal of American Veterinary Medical Association 225, 1723–1726.
Brini, E., Gandini, G., Crescio, I., Fatzer, R. and Casalone, C. (2004) Necrosis of hippocampus and piriform lobe: clinical and neuropathological findings in two Italian cats. Journal of Feline Medicine and Surgery 6, 377–381.
Cizinauskas, S., Fatzer, R., Schenkel, M., Gandini, G. and Jaggy, A. (2003) Can idiopathic epilepsy be confirmed in cats? Journal of Veterinary Internal Medicine 17, 246.
Fatzer, R., Gandini, G., Jaggy, A., Doherr, M. and Vandevelde, M. (2000) Necrosis of hippocampus and piriform lobe in 38 domestic cats with seizures: a retrospective study on clinical and pathologic findings. Journal of Veterinary Internal Medicine 14(1), 100–104.
Finnerty, K.E., Barnes Heller, H.L., Mercier, M.N., Giovanella, C.J., Lau, V.W. and Rylander, H. (2014) Evaluation of therapeutic phenobarbital concentrations and application of a classification system for seizure in cats: 30 cases (2004–2013). Journal of American Veterinary Medical Association 15, 244(2), 195–199.
Pakozdy, A., Leschnik, M., Sarchahi, A.A., Tichy, A.G. and Thalhammer, J.G. (2010) Clinical comparison of primary versus secondary epilepsy in 125 cats. Journal of Feline Medicine and Surgery 12, 910–916.
Pakozdy, A., Gruber, A., Kneissl, S., Leschnik, M., Halasz, P. and Thalhammer, J.G. (2011) Complex partial cluster seizures in cats with orofacial involvement. Journal of Feline Medicine and Surgery 13(10), 687–693.
Pakozdy, A., Sarchahi, A.A., Leschnik, M., Tichy, A.G., Halasz, P. and Thalhammer, J.G. (2012) Treatment and long-term follow-up of cats with suspected primary epilepsy. Journal of Feline Medicine and Surgery 15(4), 267–273.
Pakozdy, A., Halasz, P., Klang, A., Bauer, J., Leschnik, M., Tichy, A., Thalhammer, J.G., Lang, B. and Vincent, A. (2013) Suspected limbic encephalitis and seizure in cats associated with voltage-gated potassium channel (VGKC) complex antibody. Journal of Veterinary Internal Medicine 27(1), 212–214.
Pakozdy, A., Halasz, P., Klang, A. (2014) Epilepsy in cats: theory and practice. Journal of Veterinary Internal Medicine 28(2), 255–263.
Quesnel, A.D., Parent, J.M. and McDonell, W. (1997) Clinical management and outcome of cats with seizure disorders: 30 cases (1991-1993). Journal of American Veterinary Medical Association 210(1), 72–77.
Rusbridge, C. (2005) Diagnosis and control of epilepsy in the cat. In Practice 27, 208–214.
Schmied, O., Scharf, G., Hilbe, M., Michal, U., Tomsa, K. and Steffen, F. (2008) Magnetic resonance imaging of feline hippocampal necrosis. Veterinary Radiology and Ultrasound 49(4), 343–349.
Schriefl, S., Steinberg, T.A., Matiasek, K., Ossig, A., Fenske, N. and Fischer, A. (2008) Etiologic classification of seizures, signalment, clinical signs, and outcome in cats with seizure disorders: 91 cases (2000-2004). Journal of the American Veterinary Medical Association 233, 1591–1597.
Schwartz-Porsche, D. and Kaiser, E. (1989) Feline epilepsy. Problems in Veterinary Medicine 1, 628–649.
Timmann, D., Cizinauskas, S., Tomek, A., Doherr, M., Vandevelde, M. and Jaggy, A. (2008) Retrospective analysis of seizures associated with feline infectious peritonitis in cats. Journal of Feline Medicine and Surgery 10(1), 9–15.
Tomek, A., Cizinauskas, S., Doherr, M., Gandini, G. and Jaggy, A. (2006) Intracranial neoplasia in 61 cats: localisation, tumor types and seizure patterns. Journal of Feline Medicine and Surgery 8, 243–253.
Troxel, M.T., Vite, C.H., Van Winkle, T.J., Newton, A.L., Tiches, D., Dayrell-Hart, B., Kapatkin, A.S., Shofer, F.S. and Steinberg, S.A. (2003) Feline intracranial neoplasia: retrospective review of 160 cases (1985–2001). Journal of Veterinary Internal Medicine 17, 850–859.
Vanhaesebrouck, A.E., Posch, B., Baker, S., Plessas, I.N., Palmer, A.C. and Constantino-Casas, F. (2012) Temporal lobe epilepsy in a cat with a pyriform lobe oligodendroglioma and hippocampal necrosis. Journal of Feline Medicine and Surgery 14(12), 932–937.
Volk, H., Coleing, J.A.L., Platt, S.R. and Chandler, K. (2007) Clinical presentation and response to treatment of cats with epilepsy. Proceedings of British Small Animal Veterinary Association Congress.
Wagner, E., Rosati, M., Molin, J., Foitzik, U., Wahle, A.M., Fischer, A., Matiasek, L.A., Reese, S., Flegel, T., Matiasek, K. (2014) Hippocampal sclerosis in feline epilepsy. Brain Pathology Apr 3. doi: 10.1111/ bpa.12147.
Wahle, A.M., Bruhschwein, A., Matiasek, K., Putschbach, K., Wagner, E., Mueller, R.S., Fischer, A. (2014) Clinical characterization of epilepsy of unknown cause in cats. Journal of Veterinary Internal Medicine 28(1), 182–188.
9 Mimics of Seizure Activity: Disorders
Confused with Epilepsy
Simon Platt BVM&S MRCVS Dipl. ACVIM (Neurology) Dipl. ECVN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
Seizures are sudden onset (paroxysmal) clinical events, which represent an abnormality of forebrain neurotransmission. Epilepsy is characterized by recurrent epileptic seizures and therefore implies a less transient aetiology. The underlying abnormality can be due to structural or chemical interference with the normal ‘balance’ which exists in the brain between neuronal excitation and inhibition. Disruption of this balance may result in other clinical signs referable to forebrain dysfunction such as behavioural and visual abnormalities, but this is not necessarily the case and the affected dog or cat could be completely neurologically normal apart from the seizure activity.
Unfortunately a disturbance of the electrical circuitry in the nervous system, whether structural or functional (neurochemical), may result in other paroxysms, which can closely resemble seizure activity. These include narcolepsy, cataplexy, movement disorders (dyskinesias), sleep disorders and tremor syndromes. Further to disorders of the central nervous system (CNS), disorders of peripheral nerves and muscles, the respiratory and cardiovascular systems and systemic metabolism may result in paroxysmal ‘collapse’, which must also be considered when the possibility of a seizure event has been raised.
It is obviously extremely important to distinguish the nature of the paroxysm as this will determine the most appropriate diagnostic and therapeutic steps to take, and not least, in the case of seizure activity, immediately confirm a forebrain localization. In order to make this important determination, it is necessary to be comfortable with answering the clinical question: what is the clinical manifestation of a seizure? The answer to this question lies in the description of the event that is often only seen by the owner, as well as the circumstances that surround the onset of the event (Table 9.1). It is therefore important to know what the clinical characteristics (phenotype) of seizures typically are, so that the owner can be asked the most appropriate questions about the event (Table 9.2). These are detailed in Chapters 1 and 3.
Initial Event Description
Prior to consideration of the differentials of forebrain disease as the causes of seizure activity, the clinician should be as comfortable as possible that the event described by the owner, or witnessed first-hand, is actually a seizure rather than one of the several clinical ‘mimics’ of seizure activity (Table 9.1). This will dramatically affect the subsequent diagnostic investigation and further therapeutic interventions. This determination is based upon the phenotypic characterization of the event
Table 9.1. Paroxysmal events that could mimic an epileptic seizure.
Criteria for differentiation from seizure events
Non-epileptic Level of Flaccid or spastic Involuntary move-Possible historical Possible physical paroxysmal events Precipitating event consciousness collapse ment during event findings examination findings
Neuromuscular collapse
Movement disorder
Syncope
Sleep disorder
Narcolepsy
Cataplexy
Compulsive behaviour Vestibular event
Metabolic collapse,
e.g. hypoglycaemia
Activity/exercise
None to excitement/ activity/exercise
Exercise, excitement, cough
Sleep
Excitement/feeding
Excitement/feeding
None to environmental stimulation Variable
May be related to feeding times/ excitement
Normal unless impaired by respiratory compromise
Normal
Reduced to absent
Absent (REM sleep) and may progress to apparent wakefulness during event
Absent
Normal, if not accompanied by narcolepsy
Normal
Normal to depressed
Variable; long lasting
Often flaccid (e.g. myasthenia gravis). Can appear spastic in some cases of myopathy
Often spastic
Flaccid
Either
Usually accompanied by cataplexy Flaccid
No collapse
Usually spastic
Often flaccid. Can be spastic in some cases (e.g. hypocalcaemia)
May appear to be present when attempting to stand
Yes; exacerbated by attempts to stand No
Yes; rapid eye movements during event
No
No
No
Attempts to stand; nystagmus
No, except facial twitching in some cases of hypoglycaemia or hypocalcaemia
May be accompanied by dysphagia, dysphonia, regurgitation
May be purebred with early age onset
May be accompanied by cough, increased respiratory noise
Never occurs during periods of normal consciousness
Often purebred dog with early age onset
As for narcolepsy
Anxiety disorder
Periods of head tilt and or ataxia; head tremor; ear disease
Anorexia, depression, polyuria, polydipsia, vomiting, weight loss
May be normal or may be muscle atrophy, muscle pain and or reduced reflexes
Normal
Arrhythmia, pulse deficits, murmur, abnormal lung auscultation, cyanosis
Normal
Normal
Normal
Normal
Normal to nystagmus, head tilt, ataxia, vomiting
May be normal; weight loss Important questions about the episodic weakness, collapse or paroxysmal event Implications
| 1. What did the event look like? | It may be difficult to distinguish the event from seizure |
| activity, and some may be seizure events, but it will be | |
| important to establish that these events are not due to a | |
| metabolic or cardiovascular crisis. | |
| 2. Has this happened before? | Episodic or paroxysmal events that warrant investigation |
| should be recurrent. | |
| 3. How often has this happened? | The answer will provide insight into the progressive nature |
| of the disease and will serve as a marker for response to | |
| therapy. | |
| 4. Has it always had the same | Paroxysmal events are usually stereotypical. |
| characteristics? | |
| 5. Is the animal ‘normal’ immediately | A seizure may be followed by a period of confusion, visual |
| after these events? | dysfunction, compulsion or even aggression. |
| Neuromuscular disorders, movement disorders and | |
| syncopal events usually have no such associations. | |
| 6. Is there any type of trigger factor | Excitement or eating which often causes a loss of |
| that can be identified? | consciousness and/or collapse should prompt the thought |
| of narcolepsy/cataplexy. Several documented events occur | |
| during sleep such as the rapid eye movement sleep | |
| disorder. Exercise/excitement may be the trigger for the | |
| syndromes described in cavaliers and scotties as well as | |
| many neuromuscular diseases and hypoglycaemic-related | |
| collapse. Rarely, seizure events will be triggered by a | |
| specific noise or action. Events seen more than 8 h after | |
| last feeding may suggest hypoglycaemic collapse. | |
| 7. Is the animal normal in between | Any abnormalities described in-between the episodes could |
| the events? | indicate a structural CNS or neuropathic or myopathic |
| disease. Metabolic and cardiovascular diseases may be | |
| associated with a waxing and waning clinical course with | |
| some abnormalities detectable in between the acute events. | |
| 8. Are any other littermates known | Breed-associated events may be seen in related siblings; |
| to be affected? | however, underlying infectious diseases and toxicities |
| should not be ignored. | |
| 9. Is the animal stiff or floppy at the | Stiffness at the time of the event would often imply either |
| time of the event? | a seizure event, a movement disorder or a myopathy. |
| A floppy animal at the time of the event could also be a | |
| myopathy, but could also implicate a cardiovascular or | |
| metabolic disease. | |
| 10. Are the gums pale at the time | Mucous membrane pallor could well indicate a |
| of the event? | cardiovascular disease; however, metabolic diseases |
| such as Addison’s should also be considered. |
and historical confirmation of the potential precipitating events. Detailed questioning of the owner will be necessary (see Diagnostics section below).
Although generalized tonic-clonic seizures have a fairly unequivocal description, the recognition of a focal seizure can pose a real challenge for the clinician. For that reason
video footage obtained by the owner of the paroxysmal event can be of tremendous help. An epileptic seizure can be suspected based on:
• Presence of involuntary motor activity and/or abnormal mentation and behaviour and/or autonomic signs (salivation, urination and/or defecation). There is also notably increased muscle tone accompanying limb movement or collapse in most cases, which helps to differentiate seizures from other forms of collapse such as syncope.
The differential diagnosis for epileptic seizures is based upon the signalment, history and the neurological examination suggesting idiopathic, structural or reactive disease. The age of the patient may be very helpful in considering which combination of differentials is most likely.
Diagnosis of Epilepsy
The importance of the history
Many times the owner describes an event that gives rise to suspicion of an epileptic seizure but the animal may be normal. When faced with this situation, it is vital that the clinician asks very specific questions, which will help to determine whether the event could have been a seizure and what the underlying cause may be.
cryptogenic epilepsy. Focal seizures and focal seizures that secondarily generalize can be idiopathic in some breeds but are also associated with an intracranial disorder like structural epilepsy or cryptogenic epilepsy. Focal seizures that begin in the facial muscles and then generalize with little motor movement can be seen in cats and dogs.
disorders can appear clinically similar to seizures and can also be familial, caution should be taken with interpretation of this finding.
Neurological evaluation of a patient with paroxysmal events
In a patient with a paroxysmal event that could be seizure activity, where a neurological condition is suspected rather than abnormalities of other systems, a detailed history and neurological examination (see Chapter 10, Boxes 10.1 to 10.3) are mandatory. A thorough description of the weakness or collapse associated with the event, especially if supported by video footage, can provide important information about the speed of onset of neurological signs, potential loss of consciousness during the events and type of activity the patient was performing at the time of the ‘episode’.
The neurological examination aims to localize the lesion within the nervous system in order to list appropriate differential diagnoses and establish the further tests necessary (see Chapter 10).
Paroxysmal events to consider as possible seizure mimics
There are several broad categories of disease or abnormality that should be considered when determining whether a paroxysmal event is a manifestation of a seizure disorder or not.
These include neuromuscular disorders leading to collapse (e.g. myasthenia gravis), cardiovascular disease causing syncope, sleep-related events such as REM sleep disorder and narcolepsy/cataplexy as well as a new defined group of disorders of involuntary movement that are predominantly breed related. Obsessive compulsive disorders will also be mentioned based on their stereotypical presentations and similarities to the focal sensory seizures described in veterinary medicine. These disorders will be discussed in terms of the classical presenting signs and how they may be considered differently from epileptic disorders. Absolute confirmation of the epileptic nature can only be obtained by observing simultaneously the characteristic EEG changes and physical manifestation of the seizures.
Neuromuscular Collapse
Activity-associated weakness is the most typical clinical sign of neuromuscular disease. The interpretation of the neurological examination may be challenging in these patients. At the time of examination, they may appear normal or only mildly affected; additionally, if weakness is exhibited, it is rarely specifically indicative of nerve, neuromuscular junction or muscle disease.
In a patient with a neuromuscular disorder, observation and gait analysis may detect ventroflexion of neck, short-strided gait with over-flexion of joints (often more evident in the pelvic limbs), a plantigrade and/or palmigrade stance at rest, and generalized decreased muscle tone.
In general:
• Contrary to upper motor neuron (UMN) diseases, disorders of the lower motor neuron (LMN) do not cause ataxia, only paresis. Due to their close anatomical relationship within the caudal brainstem and spinal cord, most gait abnormalities involving the UMN pathways necessary for gait generation also cause some degree of proprioceptive ataxia. Diseases affecting the LMN system are, by definition, not ataxic. If ataxia is present in the collapsing patient, a lesion affecting the cerebellum, the vestibular system or the ascending general proprioceptive pathways in the spinal cord should be considered;
Confirming exercise/activity relationship to the problem described
Exercise testing is mandatory and represents a good tool in evaluating metabolic pathways. From rest to maximal exercise, energy demand progressively exacerbates clinical signs due to the increased muscle metabolism. The relationship of the primary complaint to activity may be obvious in some patients after a few steps but in some it may require a few minutes of activity to assess the potential effects on the gait. This is obviously very difficult in the cat. A protracted period of observation may be essential in a room where the cat cannot seek a hiding place. The main purpose of this step is to further confirm the suspicion of a peripheral neurological abnormality by determining whether the characteristics of the gait are compatible with neuromuscular disease. Additionally, this will give the observer a chance to assess the severity of the problem, the onset of pain with activity (which may not be present at rest) and the effects of activity on the cardio-respiratory systems.
Confirming whether the lesion is upper or lower motor neuron in origin
Once a neurological abnormality has been observed, the paresis must be determined to be either UMN or LMN in origin, although spinal localization is very unlikely for neurological presentation exacerbated or exclusively present at exercise. Disorders of the UMN system result in spastic paresis and normal to increased spinal reflexes, while neuromuscular conditions (LMN system disorders) are characterized by flaccid paresis, decreased muscle tone and spinal reflexes. However, many neuromuscular conditions may actually manifest with an apparent increase in tone, or stiffness, at the time of the exercise- or activity-associated weakness or collapse. Segmental spinal reflexes should be evaluated before and after exercise or activity.
Confirming whether the disease is focal versus diffuse
In a patient affected by generalized weakness associated with exercise intolerance, the neurological examination should aim to detect any other peripheral nerve dysfunctions. Diffuse neuromuscular disease often causes the spinal reflexes in all limbs to be reduced to absent. In more generalized neuromuscular diseases specific dysfunctions such as dysphagia (pharyngeal paralysis), dysphonia (laryngeal paralysis), regurgitation (oesophageal abnormalities), extraocular muscles paresis, as well as intercostal and diaphragmatic muscle weakness may be observed. Focal LMN disease may represent early onset disease especially in the acute stages, with the potential for rapid deterioration, but it may also represent a focal lesion in the spinal cord affecting the cell bodies of the LMNs.
Confirming whether the disease is affecting peripheral nerve, muscle or neuromuscular junction
From a clinical point of view, in the majority of cases, distinguishing a neuropathy from a junctionopathy or a myopathy is not possible. However, some parts of the neurological examination, including evaluation of gait, postural reactions, spinal reflexes and sensation, can be particularly helpful in distinguishing a muscular from a peripheral nerve disorder (Table 9.3). In summary:
1. Postural reaction deficits (i.e. slow hopping) are usually only present with peripheral nerve problems and not neuromuscular junction and muscle disease. Very weak patients though may be difficult to evaluate and discern whether postural reaction abnormalities are real or not.
Peripheral neuropathy Myopathy
| Mental status | Normal | Normal |
| Posture/gait | Plantigrade +/− palmigrade stance | Stiff and paretic gait |
| Flaccid paralysis | Exercise-induced weakness/stiffness | |
| Ataxia may be present if the sensory | ||
| nerve is affected (rare) | ||
| Postural reactions | Decreased (sensory) | Decreased only in severe weakness |
| Cranial nerves | May be involved | Rarely affected |
| Spinal reflexes | Decreased to absent | Decreased only in chronic muscle |
| disease | ||
| Muscle tone | Decreased (motor) | Normal to decreased |
| Sensation | Decreased to absent unless pure | Normal |
| motor nerve disease | ||
| Muscle atrophy | Present | Atrophy or hypertrophy; muscle |
| contracture may be present |
Additionally, there are some specific diagnostic tests that may help with better defining the lesion localization such as creatine kinase (CK) serum levels, electrophysiology and an edrophonium test, as presented in the ‘Specific diagnostic tests’ section below.
Differential diagnosis for neuromuscular collapse
The differential diagnoses to be considered depend on the lesion localization in addition to the signalment, history and response to previous treatment attempts. For this reason, the potential neuromuscular causes of acute generalized weakness and collapse need to be separately considered by dividing them into:
Specific diagnostic tests for neuromuscular collapse
The following tests should be considered in any case of acute neuromuscular collapse, although the diagnostic plan needs to be specifically addressed considering the suspected underlying cause (Table 9.6).
1. Rule out systemic aetiologies
i. Routine blood and urine tests: haematology, complete serum biochemistry and urine analysis should all be considered as the minimum essential database together with chest radiography and abdominal ultrasound.
ii. Serum creatine kinase evaluation: Evaluation of serum creatine kinase (CK) activity should be a part of the neuromuscular minimum database and may be indicative of active muscle disease in canine and feline patients. The serum half-life of CK is very short, lasting only 6 h; a persistent elevation of from four to five times the normal level in two tests carried out between 24 and 48 h of each other is an indication of a recent and active muscle lesion. Creatine kinase may be normal in the presence of muscle disease; therefore muscle disease should not be ruled out based on a normal CK concentration. Serum CK may also be mildly elevated in the absence of neuromuscular disease related to factors such as exercise, recumbency, trauma such as needle injections, or markedly elevated in anorexic cats. Elevations of CK are most dramatic in
Disease mechanism Examples
| Vascular | Ischaemic neuromyopathya |
| Vascular anomalies compressing the nerve roots (rare) | |
| Inflammatory/infectious | Acute idiopathic polyradiculoneuritis |
| Infectious polyneuropathies (Neospora caninum, feline | |
| leukaemia virus, feline immunodeficiency virus) | |
| Brachial plexus neuritis | |
| Toxic | Toxic/drug-induced neuritis (vincristine, |
| organophosphates) | |
| Metabolic | Diabetes mellitus |
| Hypothyroidism | |
| Electrolyte imbalance | |
| Hypoglycaemia-associated neuropathy | |
| Idiopathic | Distal denervating disease |
| Neoplastic | Neoplastic (lymphoma, peripheral nerve sheath tumour, |
| malignant sarcoma) | |
| Paraneoplastic immune-mediated polyneuropathy | |
| (insulinoma, lymphoma, pulmonary carcinoma, | |
| haemangiosarcoma) |
aIschaemic neuromyopathy usually affects pelvic limbs and is characterized by acute onset and with no progression but can infrequently be intermittent especially in dogs. It is important to assess peripheral pulses and abdominal vasculature in dogs and cats presented with intermittent weakness.
Table 9.5. Differential diagnosis in dogs (d) and cats (c) affected by acute collapse due to disorders of the muscle.
Disease mechanisms Examples
| Vascular | Ischaemic neuromyopathya |
| Inflammatory/infectious | Immune-mediated inflammatory myopathies (d) |
| Infectious polymyositis (Toxoplasma gondii, Neospora | |
| caninum, Ehrlichia spp., bacterial) | |
| Metabolic | Diabetic ketoacidosis (hypokalaemic myopathy) (c) |
| Hypothyroidism (d) | |
| Hyperthyroidism (c) | |
| Hyperadrenocorticism | |
| Hypoadrenocorticism (d) | |
| Electrolyte imbalance (hypokalaemic myopathy in cats) | |
| Hypoglycaemia (d, c) | |
| Myopathy related to glucocorticoid excess | |
| Heat stroke-rhabdomyolysis (d, c) | |
| Mitochondrial myopathy (d) | |
| Lipid storage myopathy (d) | |
| Idiopathic | Exertional rhabdomyolysis (d) |
| Breed-specific exercise-induced collapse (d) | |
| Necrotizing polymyositis (d) | |
| Neoplastic | Neoplastic (lymphoma, peripheral nerve sheath tumour, |
| rhabdomyosarcoma, metastatic neoplasia) | |
| Paraneoplastic polymyositis (lymphoma) | |
| Nutritional | Vitamin E deficiency (d) |
aIschaemic neuromyopathy usually affects pelvic limbs and is characterized by an acute in onset and static disease but can infrequently be exercise/activity related and intermittent
Table 9.6. Specific laboratory tests and associated diseases that should be considered for the investigation of neuromuscular collapse.
Laboratory tests Abnormality Suspected disease
Thyroid hormone testing
Serum glucose and serum insulin level
Cerebrospinal fluid
Serum Neospora caninum and Toxoplasma gondii titres or PCR
Plasma cholinesterase
Serum glucose level or fructosamine and glycosylated haemoglobin levels
Plasma lactate and pyruvate
Plasma, urine and muscle carnitine concentration
Serum acetylcholine receptor antibody
Serum creatine kinase (CK)
Decreased fT4, tT4 and increased TSH (d)
Albumino-cytological dissociation (increased protein associated with normal total nucleated cell count)
Elevated titres or positive PCR
Decreased plasma levels Persistent hyperglycaemia
Elevated concentrations of resting and post-exercise lactate and pyruvate
Reduced plasma and muscle carnitine, increased carnitine excretion of carnitine esters
Antibody titre > 0.6 nmol/l (d) Antibody titre > 0.3 nmol/l (c) Severely elevated CK
(100 × normal) Moderately elevated CK (10 × normal)
Hypothyroidism (hypothyroid neuropathy/myopathy) Hypoglycaemia-associated
neuropathy Insulinoma Nerve root disorders
(polyradicoloneuritis)
Neospora and Toxoplasma infection
Organophosphate toxicity Diabetes mellitus (diabetic neuropathy)
Metabolic, mitochondrial myopathies
Metabolic, mitochondrial, lipid storage myopathies
Acquired myasthenia gravis
the muscular dystrophies or with myonecrosis (100 × normal), moderately elevated in inflammatory myopathies (10 × normal), or normal or only mildly elevated in other diseases such as myotonia congenita.
iii. Radiography and ultrasound: may reveal primary cause of neuromuscular dysfunction, such as other major organ diseases, neoplasia and major blood vessel abnormalities, in addition to the consequences of NM diseases, including megaoesophagus or aspiration pneumonia.
against the ACh receptor (AChR) need to be measured in all cases of exercise-induced weakness especially if megaoesophagus is present.
2. Neuromuscular function
i. Electrophysiology: although diagnosis of a specific neuromuscular condition is rarely obtained by electrophysiology, this test is essential to classify the NM condition as a junctional, axonal, myelin or myopathic disorder and to define the distribution and severity of the disease in order to determine the differential diagnosis. This diagnostic test may also help localize the disease further to the muscle, nerve or neuromuscular junction.
ii. Muscle and nerve biopsy: necessary to provide a specific diagnosis based on electrophysiology in conjunction with clinical signs and results of other tests.
3. Specific tests Many of the tests outlined below are only performed at selected laboratories such as the Comparative Neuromuscular Laboratory at University of California, San Diego (
http: ![]() vetneuromuscular.ucsd.edu). The clinician
is advised to contact the laboratory prior to acquiring and submitting the samples for details.
vetneuromuscular.ucsd.edu). The clinician
is advised to contact the laboratory prior to acquiring and submitting the samples for details.
i. Evaluation of lactate and pyruvate levels: lactic acid is the product of anaerobic metabolism of glucose, therefore it may be increased in normal animals after anaerobic exercise. Lactic acidosis, however, can result from a defect in anaerobic metabolism, secondary to pyruvate deficiency. Plasma lactate, pyruvate and their ratio in blood are evaluated in metabolic myopathies and are essential for the diagnosis of mitochondrial diseases, characterized by high serum lactate and pyruvate concentrations with a high lactate-to-pyruvate ratio.
ii. Quantitative urinary organic acid analysis: organic acidurias can be due to inborn metabolic errors causing accumulation of non-metabolizable organic acids in tissues such as muscle and brain. Their excess can be detected in urine. Organic acidurias can result in intermittent weakness and collapse.
iii. Carnitine evaluation: primary or secondary carnitine metabolism disorders are diagnosed by complete evaluation of carnitine status in the body, through measurement of carnitine in urine, blood and muscle in its three different forms: total, free and esterified.
iv. Cerebrospinal fluid analysis: rarely useful in the evaluation of a primary neuromuscular condition; a lumbar CSF tap may be performed if a diffuse inflammatory condition affecting the nerve roots (polyradiculoneuritis) is suspected. Specific PCR for infectious agents may be performed on CSF if a diffuse inflammatory condition is suspected.
Involuntary Movement Abnormalities
Involuntary movement abnormalities result in some of the most dramatic clinical presentations in veterinary medicine. Classically, involuntary movement disorders are present during periods of inactivity rather than during voluntary movement. Some involuntary movements are persistent while others are episodic. Certain involuntary movements have characteristics that allow for identification of specific causes, whereas others are only a reflection of dysfunction of the nervous or musculoskeletal systems. Clinically, it is important first to identify the type of involuntary movement present. Subsequently, a more directed approach can be used to establish the cause of the movement disorder.
Paroxysmal events are characterized by the sudden and reversible onset of neurological dysfunction in an otherwise normal animal. Some movement disorders can be paroxysmal. The animals do not lose consciousness and rarely have a structural lesion identifiable within the CNS. The underlying cause of many of these events may be a functional abnormality related to neurotransmitter imbalances or receptor abnormalities and dysfunction. Several stereotypical events have been described in specific breeds and are discussed below. Confirmation of the specific syndrome is difficult or impossible in the clinical setting but depends heavily on the exclusion of structural CNS abnormalities such as neoplasia, inflammation and cerebrovascular disease.
Types of involuntary movements
Terms such as tics, twitches, shivering, shuddering and fasciculation are often used to describe episodic, irregular muscle movements or depolarization associated with muscle contractions (Table 9.7). Involuntary movements, however, are usually manifested through abnormal motion of the limbs, trunk or head.
Myoclonus
Myoclonus is a shock-like contraction of a muscle, or muscles, that tends to occur repeatedly in a rhythmic pattern and may persist during sleep. It is akin to the rhythmic depolarization and contraction that occurs in the heart with each beat. Myoclonus can be focal, multifocal or generalized; it often presents in a thoracic limb, however, a pelvic limb or the
Table 9.7. Definitions of most common involuntary movement manifestations.
Clinical sign or syndrome Definition
Cramp Muscle cramps are involuntarily and forcibly contracted muscles that
do not relax Dyskinesia Difficulty or distortion in performing voluntary movements Dystonia Sustained muscle contractions cause twisting and repetitive
movements or abnormal postures Fasciculation Involuntary contractions or twitching of groups of muscle fibres Myotonia Sustained muscular contraction following an initiating stimulus Myoclonus Rhythmic movement of a portion of the body resulting from sudden
involuntary contraction and relaxation of muscle groups Myokymia Continuous involuntary muscle twitching that gives the appearance of
worm-like rippling of muscle Rigidity Increased resistance to change in position or angle of joint(s) Spasm A brief, automatic jerking movement Spasticity A state of increased tone of a muscle Tetanus Sustained muscular contraction without a period of relaxation Tetany Intermittent tonic muscular contractions Tremor Any abnormal repetitive shaking movement of the body
facial muscles including the tongue may also be involved. Myoclonus may be physiological (such as that seen when falling asleep or during sleep), epileptic or symptomatic associated with central nervous system disease. An idiopathic, essential myoclonus has been recognized in people but has not been described in veterinary medicine. Myoclonus in dogs is usually the result of distemper infection, which establishes a pacemaker-like depolarization of local motor neurons; however, it has been associated with lead toxicity and other causes of CNS inflammation.
Myoclonus is often described to originate from spinal disease causing a localized persistent movement abnormality due to abnormal lower motor neuronal discharges (Podell, 2004). This usually affects one or two limbs and occasionally the jaw. The muscle contractions occur rhythmically and are most obvious in resting animals; they are present throughout activity and do not disappear during sleep. It can also arise from apparent cerebral disease as a type of seizure.
Tremor
Tremor is one of the most common involuntary movement disorders in humans, and is also surprisingly common as a clinical abnormality in dogs. Tremor is an involuntary rhythmic, oscillating movement of fixed frequency resulting from alternate or synchronous contraction of reciprocally innervated antagonistic muscles (Jankovic and Fahn, 1980). It can be focal, affecting just one limb or the head for example, or generalized. Electromyographically, tremor is characterized by rhythmic bursts of motor neuron activity occurring in opposing muscle groups. The contraction of muscles with opposing function gives tremor a biphasic nature. This biphasic character differentiates tremor from other abnormalities of movement. While seen during the awake state, true tremor should cease with sleep. As for myoclonus, tremors may be physiological, idiopathic (or essential) such as that seen in senile tremor of dogs, or pathological due to a nervous system disease.
Tremor is ultimately a disorder of movement (Jankovic and Fahn, 1980). Therefore, lesions in any of the regions of the central and peripheral nervous systems and musculoskeletal system primarily responsible for normal movement may generate a tremor. This makes localization challenging when considering the clinical signs alone. In humans, important motor areas include the basal nuclei and other components of the extrapyramidal system, the cerebellum, diffuse neuronal cell bodies involved in segmental and supraspinal reflex mechanisms, components of the lower motor neuron and the interconnecting pathways. Additionally, abnormalities of the mechanical apparatus of the limbs (e.g. bones, joints and tendons) may also result in tremor as a result of pain and weakness. However, species differences do exist, and it is important to note that lesions involving the basal nuclei and substantia nigra commonly result in tremor in human beings but not in dogs.
Tremors that occur or worsen when an animal is trying to perform purposeful movements (intention tremors) are most often associated with cerebellar disease. Fine tremor (decreased amplitude and increased frequency) is more often associated with diffuse neuronal disease or muscle weakness (Garosi et al., 2005). The causative lesion may give rise to other signs of neurological dysfunction that can help further define the localization, such as dysmetria associated with cerebellar disease.
Myokymia and neuromyotonia
Myokymia and neuromyotonia refer to involuntary rippling of muscles that persists even during sleep and under anaesthesia (Vanhaesebrouck et al., 2010b; Bhatti et al., 2011). Myokymia is one of the clinical signs of neuromyotonia. Neuromyotonia is clinically characterized by a combination of muscle twitching or myokymia, persistent muscle contraction, muscle stiffness or cramps and impaired muscle relaxation (Bhatti et al., 2011). Axonal voltage-gated potassium channel abnormalities may be responsible for the condition secondary to autoantibody damage, toxicity or genetic mutations. This condition has been sporadically reported in dogs and cats but is most well documented in Jack Russell terriers (Van Ham et al., 2004; Galano et al., 2005; Walmsley et al., 2006; Vanhaesebrouck et al., 2010a, b; Bhatti et al., 2011).
Electromyography in myokymia reveals short bursts of ectopically generated motor unit potentials, firing at rates of 5–62 Hz and appearing as doublets, triplets or multiplets. These bursts fire rhythmically or semi-rhythmically, and sound like soldiers marching. Neuromyotonia is characterized by muscle stiffness and persistent contraction related to underlying spontaneous repetitive firing of motor unit potentials. On EMG there are prolonged bursts of motor unit potentials, firing at rapid rates of 150–300 Hz, which begin and end abruptly, do not occur repetitively in a rhythmic fashion and have characteristic waning amplitude.
Fasciculations arise from ectopic electrical activity in the distal axon and are typically the manifestation of irritability of the neuronal cell body or its associated axons (Podell, 2004).
The clinical and clinicopathological findings, treatment and outcome of myokymia and neuromyotonia in 37 Jack Russell terriers was recently reported (Bhatti et al., 2011). The most characteristic clinical signs were episodes of rhythmic, undulating muscle contractions that induced vermicular movements of the overlying skin. Collapse and recumbency were also seen in these dogs, which exhibited rigidity. The episodes were mostly triggered by excitement, exercise or hot weather. Electrophysiological investigation can reveal neuromyotonic discharges, which characteristically are represented by semi-rhythmic bursts of doublet, triplet or multiplet discharges of a single motor unit (Vanhaesebrouck et al., 2010b).
Drugs that cause a membrane stabilizing effect on all cell membranes, including those of the peripheral nerves, have been effectively used; these include the sodium-channel blockers procainamide and mexilitine which have been used in dogs as has slow-release phenytoin with variable effect (Bhatti et al., 2011). Cold water baths have been more uniformly successful especially when combined with general anaesthesia.
Fasciculations arise from ectopic electrical activity in the distal axon and are typically the manifestation of irritability of the neuronal cell body or its associated axons (Podell, 2004).
Dyskinesia
Dyskinesia is defined as impairment of the power of voluntary movements resulting in fragmented or incomplete movements (Ramsey et al., 1999; Penderis and Franklin, 2001). Dogs reported with these abnormalities may exhibit abnormal postures such as holding up a limb in an attempt to move or adopting a kyphotic posture of the spine without being able to initiate movement. The pathophysiologic mechanisms underlying these movements are poorly understood, but may represent a central neurotransmitter or pathway abnormality, or possibly a local muscular abnormality. The impaired movement can appear as and have been termed muscle ‘cramps’, which are defined as paroxysmal, prolonged and severe contraction of muscles that may be painful and can be either focal or generalized (Shelton, 2004). Examples of diseases associated with cramps which may be dyskinesias include Scotty Cramp, Episodic Falling of Cavalier King Charles spaniels, ‘Epileptoid cramping’ of Border terriers, and extreme generalized muscular stiffness in male Labrador retrievers (Vanhaesebrouck et al., 2011). Muscle cramps have also been described secondary to systemic diseases such as hypoadrenocorticism.
Dyskinesias are movement disorders that occur spontaneously during activity or at rest causing involuntary contractions of groups of muscles in a conscious animal. The descriptions of these conditions indicate that the most common clinical sign is that of dystonia causing increased muscle tone in one or several limbs, possibly leading to collapse. The movements can be triggered by excitement or exercise. The localization of the purported functional neurotransmitter-based abnormalities responsible for these disorders may be the central or peripheral nervous system. In general, movement disorders may have origins in the cerebrocortical neurons, basal nuclei or peripheral nervous system (Packer et al., 2010).
Diagnostic approach for involuntary movement disorders
The clinical presentation of movement disorders is complex, often variable and sometimes even bizarre. Establishing the correct diagnosis can, therefore, be difficult. Obtaining an accurate history of the patient is important to define the onset and progression of the condition in addition to elucidating any underlying systemic health problems that could be causing the disorder.
Physical examination is essential as some tremor disorders may be associated with systemic disease. Many tremor syndromes may also be associated with neurological deficits; therefore a neurological examination can help to localize the causative lesion or associated deficits and determine the next stages necessary in the diagnostic work-up.
The following tests should be considered in most patients with movement disorders:
Establishing the aetiology using clinical characteristics
The clinical characteristics of the abnormality may not only suggest that it is a movement disorder rather than one of the many mimics but may aid in the underlying aetiology. Various classification schemes have been proposed based on the presence of activity at the time of the disorder, whether they are continuous or episodic, involvement of muscle or nerves and whether there is too much or too little movement. Ultimately, these schemes can be overcomplicated in veterinary medicine when so few of these disorders are seen and definitively diagnosed. More simply, the abnormalities can be investigated by dividing them into localized or generalized syndromes.
Localized tremor syndromes
Localized limb tremors/myoclonus
Recent classification schemes have suggested calling tremors a form of myoclonus (often action-related indicating they are more pronounced with activity). In this text, tremors and myoclonus are kept as separate entities with myoclonus referring to rhythmic activity of large groups of muscles causing flexion and extension of limbs as opposed to diseases causing more ‘fine’ movement abnormalities of small muscle groups (tremor).
There are many different causes of limb tremors and it should be remembered that focal seizures can cause involuntary movements of a single limb. Some specific diseases are described below.
• Spinal disease: tremor can occur in one limb or body area. Tremor restricted to only the pelvic limbs may be seen in dogs with lumbar and sacral disease. This tremor may result in part from muscle weakness secondary to spinal cord or peripheral nerve impingement, or possibly occurs as the reflection of pain. Pelvic limb tremor may result from compressive diseases such as lumbosacral vertebral canal stenosis, neoplasia and discospondylitis;
Localized head tremors
Dogs occasionally have tremor involving only the head. This type of tremor most likely results from tremor of the neck muscles but its pathophysiology is poorly understood. Head tremors which are exacerbated by an intentional movement such as eating or drinking are termed intention or ataxic tremors. This abnormality indicates cerebellar dysfunction. Paroxysmal or continuous non-ataxic tremors of the head are often considered to result from cerebral or thalamic diseases. Focal facial movement abnormalities or intermittent head movements/jerks should also be considered as potential seizure disorders and investigated appropriately.
Non-ataxic head tremors
head bob as their only clinical sign can have an inflammatory or infectious disease (see Chapter 5 for a description of the different causes of encephalitis). In addition, myoclonus of the head or face can be seen with inflammatory diseases of the central nervous system. This may be more common with distemper virus infections and has been called ‘chewing gum fits’ due to the rhythmic jaw movements seen clinically. However, it may be difficult to distinguish this form of localized myoclonus from continuous focal seizure activity. Focal facial movement abnormalities or intermittent head move-ments/jerks should always be considered as potential seizure disorders and investigated appropriately.
Ataxic head tremors
Tremors that occur when an animal intends to move in a goal-oriented activity are most often the result of cerebellar disease. This tremor may involve the whole body but is usually most obvious in the head. The head usually moves in an up and down (‘Yes’) direction at a frequency of 2–4 Hz. This type of tremor is exaggerated by goal-oriented movement such as eating. This is most likely a dysmetria of head movement.
Generalized tremor syndromes
Generalized tremors and involuntary movements are surprisingly common in dogs (Wagner et al., 1997). Neurolocalization can be difficult as they could result from focal vestibulo-cerebellar diseases or more diffuse central or peripheral nervous system diseases. These events can result secondary to intoxications, drug therapies, congenital myelin abnormalities, storage diseases, encephalitis, hypertension, vascular disease or may arise without a definable cause. These underlying aetiologies can be ruled out with a good history, a minimum database, a CSF tap and advanced imaging. When all the tests are normal consideration should be given to whether the syndrome is a tremor in which case it may be idiopathic in nature (essential tremors) or whether it is a paroxysmal onset of abnormal muscle tone and movement, in which case one of the several breed-related ‘dyskinesias’ should be considered.
Essential tremors/geriatric (senile) canine tremors
Physiological and essential tremors are most common in people. Essential tremors are considered exaggerated forms of physiological tremors and when they occur in later life they are termed senile tremors. Some older dogs will exhibit a fine tremor of the pelvic limbs as they age and the condition can be slowly progressive. It is a postural related tremor and as such is only present when the dog is standing. There is no effect on strength or gait in these dogs and no pain is detected. No treatment is necessary unless symptomatic therapy is required to improve a perceived quality of life issue.
Paroxysmal dyskinesias
Paroxysmal dyskinesias are episodes of abnormal involuntary hyperkinetic movement or muscle tone. These events are distinguished from seizures by the presence of a normal consciousness, although an EEG would be necessary definitively to determine this.Amovement disorder has been described in young bichon frisé dogs with an extreme variability of frequency and random occurrence (Penderis and Franklin, 2001). A rapid muscular contraction causes hyperflexion and/or extension of an individual limb. The thoracolumbar spinal column can be affected by altered muscle tone during the event causing a kyphotic posture. A similar condition has also been described in young boxer pups provoked by excitement causing abnormal facial, truncal and limb movements with sustained hyperflexion (Ramsey et al., 1999).
No successful treatment regimens have been described. It remains to be seen whether a genetic disorder confirms these as truly breed-related disorders as documented below. Several drugs have been reported to cause similar dyskinesias and include phenobarbitone and propofol in dogs (Smedile et al., 1996; Kube et al., 2006). These disorders are usually reversible with drug tapering or withdrawal.
Scotty cramp
Clinical episodes of dystonia are most commonly seen in Scottish terriers from 6 weeks to 3 years of age and may be elicited by stress, excitement or exercise. The thoracic limbs are initially affected, becoming abducted shortly after exercise begins; this is followed by arching of the lumbar spine and pelvic limb stiffness, which can progress to somersaults, falling, and tightly flexed pelvic limbs. Loss of consciousness is not a feature and the signs resolve within 10 min, but can recur multiple times over a 24-h period. Similar conditions have been described in dalmatians, a cocker spaniel, a wirehaired terrier, wheaton terriers, Norwich terriers and Border terriers. In Border terriers the disease has been termed ‘Spikes’ disease or canine epileptoid cramping syndrome (CECS). A breeder-run website contains further information on this condition (
http:![]() www.borderterrier-cecs.com).
www.borderterrier-cecs.com).
This recessively inherited non-progressive disorder is thought to be associated with relative deficiencies of the inhibitory neurotransmitter 5-hydroxytryptamine (serotonin).
A presumptive diagnosis is based on clinical signs and breed. All laboratory tests are within normal limits. Signs can be induced with exercise 2 h after using methylsergide
(0.3 mg/kg orally), a serotonin antagonist.
Treatment consists of daily oral dosing of acepromazine maleate (0.1–0.75 mg/kg q12h) or diazepam (0.5 mg/kg q8h). Vitamin E (125 IU/kg/day) has also been advised for these dogs. Serotonin reuptake inhibitors such as fluoxetine may be useful in affected dogs. Non-steroidal anti-inflammatories are contra-indicated. Prognosis is fair, as the disease is non-progressive; appropriate lifestyle changes can result in a good quality of life. In CECS, dietary treatment using hypoallergenic foods has been suggested but no evidence exists for this at this time.
Episodic hypertonicity in cavalier King Charles spaniels
This condition, also known as ‘episodic falling syndrome’ in cavalier King Charles spaniels,
has been described in the UK, USA and Australia and is suspected to have an inherited component. The genetic locus for this condition has recently been mapped to canine chromosome 7 with approximately 13% of the breed suggested to be carriers for the disease (Gill et al., 2012).
The syndrome is often seen in animals between 3 and 7 months of age but can affect animals up to 4 years of age. Variable periods of exercise induce a bounding pelvic limb gait in which the limbs may be abducted and appear stiff. This may progress to ‘bunny-hopping’, arching of the spine and collapse. As in Scotty cramp, the animals are normal between the events, there is no loss of consciousness and the events may be triggered by exercise, stress and excitement.
The pathogenesis is at present unknown but preliminary studies implicate an abnormality of CNS neurotransmission. Recent work has identified a genetic deletion affecting the brevican gene in affected dogs and confirming the disease as an autosomal recessive (Gill et al., 2012). Brevican belongs to a family of aggregating extracellular matrix proteoglycans where it has a role in governing synapse stability; it is highly expressed in the CNS.
Laboratory tests and electrodiagnostic examinations are normal. Therefore diagnosis used to be by exclusion and correlation of an appropriate history with clinical signs. However, a genetic test is now available for the confirmation of this disease.
Treatment with the benzodiazepine drug clonazepam (0.5 mg/kg q8h) can result in almost complete remission of the signs but tolerance to this drug does develop. The carbonic anhydrase inhibitor acetazolamide may have therapeutic benefit in these dogs.
Startle disease in Irish wolfhounds
Hyperekplexia or startle disease is characterized by noise- or touch-induced non-epileptic seizures that result in muscle stiffness and apnoea in people (Gill et al., 2011).
Defective inhibitory glycinergic transmission due to genetic mutations affecting the glycine receptor is usually the cause. It has been suggested that familial reflex myoclonus in Labrador retrievers could be a glycinergic transmission disorder and represent a startle disorder. A startle disease has recently been documented in Irish wolfhounds in the USA with a microdeletion in the gene encoding a presynaptic glycine transporter (Gill et al., 2011). The condition was seen to develop in 5–7-day-old pups, evoked by handling and abating when relaxed or sleeping. The puppies affected could not stand and had a rigid posture in all four limbs with generalized tremor. Progressive feeding difficulty was noted and euthanasia was performed by 3 months of age. Carriers of this disease can now be identified.
Generalized muscle stiffness in male Labrador retrievers
Young male Labradors have been described with a paroxysmal generalized rigidity of CNS origin. Signs have been seen to start between 2 and 41 months of age, with a mean age of onset being 17 months, stabilizing in adulthood (Vanhaesebrouck et al., 2011). The disease seems first to affect the pelvic limbs and then progresses to the thoracic limbs causing them to present for exercise intolerance. All affected dogs seem to exhibit generalized muscle stiffness, persisting at rest and resulting in restricted joint movements. The dogs have a flexed posture and a bradykinesia or extreme slowness of movements and reflexes.
Currently it is thought that this condition arises from basal nuclei and reticular formation abnormalities together with motor neuron disinhibition caused by a decreased number of spinal cord interneurons. Initial pedigree analysis suggests an X-linked hereditary disease.
Conscious needle EMG showed continuous motor unit activity in the proximal limb and epaxial muscles while the dogs were standing or laterally recumbent (Vanhaesebrouck et al., 2011). Serum creatine kinase levels are normal as is the CBC, serum chemistry and CSF. Therefore diagnosis is by exclusion and correlation of an appropriate history with clinical signs.
A poor quality of life can lead to requests for euthanasia. Clinical signs can progress but have been seen to stabilize. Treatment with nonsteroidal anti-inflammatory drugs have been shown to provide partial and temporary improvement in some dogs however no specific and uniformly successful treatment is known.
Paroxysmal dyskinesia in Chinooks
A paroxysmal dyskinesia has been described in related chinooks characterized by an inability to stand or ambulate, head tremors and involuntary flexion of one or multiple limbs, without autonomic signs or loss of consciousness (Packer et al., 2010). Episode duration varies from minutes to 1 h. Based on pedigree analysis, the disorder was considered to be consistent with a partially penetrant autosomal recessive or polygenic trait. There has been some consideration given to whether this disorder is an atypical seizure episode but the same can be said for any of the aforementioned dyskinesias.
Dancing doberman disease
A condition affecting dobermans and causing intermittent flexion of one or both pelvic limbs has been reported. Muscle atrophy, weakness and postural reaction deficits may be seen in chronic cases. The gastrocnemius muscle appears to be the primary focus of the disease with EMG changes detectable including positive sharps, fibrillation potentials and complex repetitive discharges. The underlying cause is not known but sciatic-tibial nerve biopsy may reveal axonal disease, which may be primary or secondary in origin.
Syncope
The term syncope, from the Greek for ‘cutting short’, refers to an abrupt and transient loss of consciousness accompanied by loss of muscular tone. It is usually caused by a sudden, global reduction in cerebral perfusion, and clinical recovery occurs with restoration of normal cerebral blood flow. The very transience of this syndrome and the variety of medical disorders that can cause or mimic it are at the core of the diagnostic problems that the neurologist faces. The term ‘fainting’ is often used synonymously with syncope and captures the essential criteria of the collapse – loss of consciousness and muscle tone.
During a syncopal event, the animal usually collapses into lateral recumbency. Stiffening of the limbs, opisthotonic posture, micturition and vocalization are common but facial ‘spasms’, persistent tonic/clonic motion, defecation, a prodromal aura, (post-ictal) dementia and neurologic deficits are not usually associated with cardiovascular syncope; however, profound hypotension or asystole can cause hypoxic ‘convulsive syncope’, with seizure-like activity or twitching. Convulsive syncopal episodes are preceded by loss of muscle tone; however, seizure activity caused by underlying neurologic disease is usually preceded by atypical limb or facial movement or staring spells before the loss of postural tone (Johnsrude, 2000). ‘Presyncope’, where reduced brain perfusion, or substrate delivery, is not severe enough to cause unconsciousness, may appear as transient ‘wobbliness’ or weakness, especially affecting the pelvic limbs.
Pathophysiology of syncope
Mechanisms underlying syncope usually involve either acutely reduced cardiac output (often related to arrhythmias, decreased cardiac filling), outflow obstruction, hypoxia or hypoglycaemia with normal cerebral blood flow, or decreased vascular resistance related to neurocardiogenic reflexes. A fall in cardiac output or vascular resistance reduces mean arterial pressure and, consequently, cerebral perfusion. Syncope occurs when cerebral blood flow falls below a critical level (30–50% of normal in people) (Johnsrude, 2000).
Reduced cerebral blood flow can also result from cerebrovascular or other intracranial disease. Syncope in dogs and cats is often associated with excitement or exertion, when the demand for cardiac output and oxygenation is increased (Calvert et al., 1996; Miller et al., 1999; Bright and Cali, 2000; Schnipper and Kapoor, 2001). Cardiac arrhythmias, as well as organic heart disease, are commonly involved. Tachyarrhythmias, such as paroxysmal ventricular or supraventricular tachycardias and atrial fibrillation, can markedly reduce cardiac output by compromising ventricular filling time and stroke volume.
Bradyarrhythmias (e.g. complete AV block, sinus arrest) can profoundly reduce heart rate and, therefore, cardiac output. Underlying cardiac functional or structural abnormalities exacerbate the negative effect of arrhythmias on cardiac output. Even when the heart rhythm is normal, diseases that cause poor myocardial contractility, impaired filling or outflow obstruction may prevent sufficient rise in cardiac output to meet increased demand during activity or excitement (Johnson et al., 1999; Ware and Hopper, 1999).
In animals with normal cardiac output, insufficient cerebral oxygen delivery can occur when blood oxygenation is impaired, as with right-to-left shunts, anaemia or severe pulmonary disease. Hypoglycaemia can also precipitate syncope, especially with exertion, although weakness and seizures are more common manifestations. Diseases that increase intracranial pressure can cause inadequate blood flow to the brain by reducing cerebral perfusion pressure and compressing intracranial vessels:
blood flow = perfusion pressure/vascular resistance; perfusion pressure = MAP – intracranial pressure.
Cerebrovascular disease can critically reduce cerebral blood flow either by vascular obstruction or rupture. Neurocardiogenic reflex mechanisms appear to cause syncope in some animals, but this is much less common than in people (Grubb, 1999; Arthur and Kaye, 2001). Quadrupedal, rather than upright, posture makes animals less susceptible to gravitational effects on the circulation and orthostatic hypotension. Neurocardiogenic (previously called vasovagal) syncope is not well-documented in animals, but syncope that occurs during sudden bradycardia after a burst of sinus tachycardia has been observed in a number of dogs, especially small breed dogs with advanced AV valve disease; excitement often precipitates such an episode. Doberman pinschers and boxers may experience a similar syndrome (Calvert et al., 1996). Whether, and to what degree, hypotension occurs in these dogs is unknown because of the difficulty in documenting blood pressure during the syncopal episode. It is suspected that an acute sympathetic surge induced by excitement or exercise provokes a strong reflex vagal response that results in bradycardia as well as hypotension. Activation of ventricular mechanoreceptors by forceful contractions, especially when ventricular filling is reduced, may play a role. The resulting surge in afferent neural discharge is thought to mimic that associated with hypertension, stimulating a paradoxical brainstem response of sympathetic withdrawal and vagal activation (Grubb, 1999). Syncope in dogs with phaeochromocytoma may represent a neurocardiogenic response to surges in sympathetic activity. It is also possible that neurocardiogenic mechanisms may be involved in animals with anaemia and syncope. Syncope precipitated by coughing (cough syncope, ‘coughdrop’) is a form of syncope that occurs more often in dogs with brachycephalic conformation, underlying airway disease or collapse, or with chronic mitral regurgitation and marked LA enlargement. Coughing transiently increases intrathoracic pressure, which reduces venous return to the heart, and intracranial pressure, which reduces cerebral per-fusion pressure. The fall in cardiac output and cerebral perfusion pressure can reduce cerebral blood flow below the level needed to maintain consciousness. Coughing can also reflexly stimulate vagally-mediated bradycardia and vasodilation, which can contribute to hypotension and syncope. The true incidence of syncope in dogs and cats is unknown.
While syncope may be under-reported, the low prevalence in dogs and cats compared with people (estimated as high as 30–50%) suggests that neurocardiogenic syncope, especially orthostatic hypotension, is rare in quadrupeds (Goodwin, 1998; Schnipper and Kapoor, 2001). Syncope occurs more frequently in older animals and is often associated with cardiac and, to a lesser extent, other disease. About two-thirds of the dogs with syncope also have cardiac disease and/or arrhythmia. Less frequently, respiratory diseases, anaemia or various other metabolic, neoplastic or neurologic conditions are concurrently diagnosed in these dogs with syncope. Similarly, about two-thirds of cats with syncope are noted to have myocardial disease and/or an arrhythmia.
Approach to the patient with syncope
Box 9.1. Aetiological classification of syncope.
The clinical history and physical examination often give clues to the underlying cause of episodic collapse (Box 9.1) (Arthur and Kaye, 2001; Schnipper and Kapoor, 2001). Detailed description of the episodes themselves, as well as preceding events, prodromal signs and the animal’s mentation and behaviour after the event, can be helpful in differentiating cardiovascular syncope from seizure activity or other causes of collapse. Other information to be collected includes the number and frequency of previous events, whether the patient has had signs of cardiopulmonary or other systemic disease, what medications the animal is taking and whether there has been collapse or sudden death in related animals. The physical examination should evaluate all body systems thoroughly, with particular focus on the cardiovascular, nervous and respiratory systems. A routine database of CBC, biochemical profile, urinalysis, heartworm test and arterial blood pressure measurement should be done. Although these tests are often normal, contributory underlying disease may be revealed. Endocrine tests (e.g. for adrenal or thyroid function) may be useful in some cases. A baseline ECG is recommended. Although a resting ECG may be non-diagnostic, it may suggest underlying cardiac enlargement, conduction abnormalities or arrhythmia that could contribute to syncope. Thoracic radiographs are taken to evaluate the lungs, pleural space, mediastinum and pulmonary vasculature, as well as the cardiac size and shape. Suspicion of an underlying cardiac cause for syncope is usually generated by the combination of history, physical examination, the ECG and thoracic radiographs. Echocardiography can confirm the presence and severity of cardiac structural or functional abnormalities that could lead to syncope or be risk factors for arrhythmias. Ambulatory ECG monitoring (e.g. with a Holter or event monitor) can help identify or exclude cardiac arrhythmias as a cause for syncope in some animals (Goodwin, 1998; Miller et al., 1999; Arthur and Kaye, 2001; Schnipper and Kapoor, 2001). The reported diagnostic yield, both positive and negative results, has ranged from 42 to
1. Cardiovascular:
i. Arrhythmias
ii. Impaired forward cardiac output
iii. Impaired cardiac filling
2. Non-cardiac:
3. Reflex:
i. Neurocardiogenic (vasovagal)
ii. Situational
iii. Carotid sinus hypersensitivity
85% (Goodwin, 1998; Miller et al., 1999; Bright and Cali, 2000). Event monitors, which are generally worn for a 1–2 week period, have a higher diagnostic yield than Holter monitors, especially in animals with structural heart disease (Bright and Cali, 2000; Arthur and Kaye, 2001; Schnipper and Kapoor, 2001). A syncopal episode must occur during monitoring to make a definitive diagnosis. While a brady- or tachyarrhythmia may underlie the syncopal event, in many cases cardiac arrhythmia can be excluded as the precipitating cause. Arrhythmias often occur without clinical signs; not all arrhythmias cause sufficient haemodynamic compromise to induce syncope or weakness (Miller et al., 1999; Ware, 1999; Bright and Cali, 2000; Calvert et al., 2000; Arthur and Kaye, 2001; Meurs et al., 2001). Holter monitoring for 24 (to 48) h is most likely to be diagnostic in animals with multiple syncopal episodes over a short period of time, although the frequency of syncope does not predict the likelihood of an event during Holter monitoring (Miller et al., 1999). Holter monitoring is useful for quantifying the type and severity of arrhythmias, for identifying arrhythmias in asymptomatic patients and for assessing antiarrhythmic drug efficacy. Continuous loop event monitors allow a longer monitoring period than Holter monitors and are better suited for patients with infrequent symptoms (Goodwin, 1998; Bright and Cali, 2000; Arthur and Kaye, 2001). These digital loop recorders monitor heart rhythm continuously; when activated, the ECG is saved into memory for a brief period prior to and following activation. The ECG data are then transmitted by telephone for printing and interpretation. The disadvantages of event monitors are that they do not record potentially significant arrhythmias unless activated, and they do not quantify the frequency of arrhythmias. Implantable loop recorders (Reveal, Medtronic) have also been used in veterinary patients with recurrent but infrequent and unexplained syncope (Willis et al., 2003). These subcutaneously implanted devices must be activated at the onset of symptoms in order to save ECG data.
Treatment of syncope
Therapy is aimed at managing underlying disease and avoiding precipitating factors, such as exertion or environmental stressors, as far as possible. This may include instituting or adjusting medications for heart failure or hypertension correcting anaemia, or treating respiratory or metabolic diseases. When an arrhythmia appears to be the cause, appropriate antiarrhythmic drug therapy or pacing is indicated but pacing is unlikely to modulate hypotension caused by neurocardiogenic syncope. Several strategies have been tried to manage suspected neurocardiogenic syncope. Beta-blockers can be used as a means to blunt the initiating sympathetically-induced tachycardia and/or vigorous ventricular contraction, but exacerbation of bradycardia can be a concern. Other strategies that have been effective anecdotally include theophylline or aminophylline, starting with a low dose and titrating up to effect, or low-dose digoxin in animals with AV valve disease.
Narcolepsy/Cataplexy and Sleep Disorders
Narcolepsy is a disorder of sleep/wake control characterized by a tendency to fall asleep during the day, disturbed night-time sleep patterns and cataplexy (Dauvilliers et al., 2007; Nishino, 2007; Tonokura et al., 2007).
Cataplexy refers to sudden loss of motor tone ranging in severity from a dropped jaw to complete collapse without loss of consciousness and it represents a disorder of rapid eye movement (REM) sleep. Narcolepsy has been reported in many canine breeds, including the Doberman pinscher, Labrador retriever, miniature poodle, beagle and dachshund.
The predominant sign in dogs and cats is cataplexy but excessive daytime sleepiness and fragmented sleep patterns have also been reported. Cataplexy is characterized by paroxysmal attacks of flaccid paralysis without loss of consciousness and may last up to 20 min, with a sudden return to normality. The event is not accompanied by faecal or urinary incontinence, salivation or rigidity of muscle groups. The episodes, which may occur multiple times a day, are frequently induced by excitement such as eating or playing and they can be reversed by verbal or tactile stimuli. Cataplexy has been recorded in puppies and adult dogs but usually begins in the first 6 months of life with the establishment of REM sleep.
The pathogenesis of this disorder remains uncertain; however, an imbalance between cholinergic and catecholaminergic neurotransmitter systems within the CNS appears to be involved. Recent studies point to hypocretins (orexins) as important sleep-modulating neurotransmitters (Wu et al., 2002, 2011; Mishima et al., 2008; Kroeger and de Lecea, 2009). The hypocretins (orexins) are two novel hypothalamic neuropeptides (Hcrt-1 and Hcrt-2), derived from the same precursor gene, that are synthesized by hypothalamic neurons. Hypocretin secretion increases during wakeful states and decreases during sleep. Defects in hypocretin neurotransmission and hypo-cretin deficiency appear to play an important role in narcolepsy (Lindberg et al., 2007). Inherited forms of canine narcolepsy have a hypocretin receptor abnormality and sporadic forms have decreased CSF hypocretin concentrations (Tonokura et al., 2003; John et al., 2004). Autoimmunity is additionally considered by some researchers to play a role in the development of narcolepsy, in view of the rarity of hypocretin receptor mutations in humans and a genetic link to a human leucocyte antigen (HLA) locus. However, such an association has not been found in dogs and immunosuppressive treatment is not effective. Autosomal recessive inheritance with full penetrance was established in Doberman pinschers and Labrador retrievers and linked to a region of chromosome 12 that was termed the canarc-1 gene in both breeds. Subsequent work has shown that there are mutations in the hypo-cretin (orexin) receptor 2 gene (Hcrtr2) in this region. Dogs with the inherited form have normal CSF levels of hypocretin 1. A familial form exists in the dachshund.
Diagnosis is based on typical clinical signs, though routine analysis of CSF hypocretin levels may become available in the future.
Attacks can be induced in most affected animals by exercise or eating. Signs can be alleviated for up to 45 min using imipramine
(0.5 mg/kg IV). Atropine sulfate (0.1 mg/kg IV) is also reported to be a useful diagnostic test, providing immediate temporary remission of signs for up to 3 h.
The disease may not be progressive with respect to frequency or severity of the events but it can obviously affect the animal’s quality of life. Long-term treatment with tricyclic antidepressants such as imipramine hydrochloride (2–4 mg/kg orally q8–12h) and desipramine (3 mg/kg orally q12h) have been recommended based on their inhibition of norepinephrine reuptake in the brain. Methylphenidate hydrochloride has also been described as effective at a dose of 0.25 mg/kg q12–24h (Mignot, 2004). Venlafaxine has recently been reported as a potential treatment for narcolepsy in dogs (Delucchi et al., 2010).
Hypocretin replacement therapy has been attempted but without much success at this time (Fujiki et al., 2003; Schatzberg et al., 2004; Brisbare-Roch et al., 2007).
Rapid eye movement sleep disorders
Normal sleep is divided into two stages called non-rapid eye movement, the first stage of sleep lasting 20 min, and rapid eye movement (REM) sleep (Schubert et al., 2011). During non-REM sleep there is a decrease in body temperature, heart rate and respiratory rate and the animals are immobile but retain muscle tone. REM sleep lasts for about 15 min during which animals have an increase in body temperature, heart rate and respiratory rate coincident with the eye movements and atonia of the postural muscles. Normal movements seen during this phase can include twitching of the eyelids, face, larynx and paws with occasional rhythmic paddling of all four limbs and yelping.
The cerebral cortex is highly active during REM sleep and if the lower motor neuron ventral horn cells in the brainstem and spinal cord are not inhibited as occurs normally, uncontrolled limb movements will be seen (Boeve et al., 2007a, b). Normally the pons and medulla control this inhibition and any disease of these structures could result in REM sleep abnormalities.
REM sleep behaviour disorder has recently been documented in dogs (Bush et al., 2004; Schubert et al., 2011). Almost 30% of dogs in one study were golden retrievers. The episodes were seen mostly in young dogs (64% were less than 1 year old) but affected dogs from 2 months to 7.5 years old. Violent limb movements, howling, barking, growling, chewing or biting during sleep is seen in most affected dogs. Most dogs can be easily aroused and awoken to be completely alert.
The diagnosis can be made based on clinical signs but confirmation depends on EMG and EEG studies during the event, which is logistically difficult in veterinary medicine.
Treatment with oral potassium bromide at 40 mg/kg/day reduced the severity and frequency of events in nearly 80% of dogs. Phenobarbital appears to have minimal effect on this disorder in dogs but tricyclic antidepressants such as amitriptyline may be helpful. Spontaneous recovery has not been reported.
Compulsive Behavioural Disorders
In dogs and cats, behaviours such as ‘fly biting’ and tail chasing have commonly been considered symptomatic for seizure disorders, although treatment with anti-epileptic medications may not be successful. These abnormal behaviours in companion animals have also been considered homologous to the stereo-typic behaviour of livestock and zoo animals (Luescher et al., 1991). Such behaviours share similarities with human obsessive compulsive disorder (OCD) and have been referred to as OCD or compulsive disorders (CD) (Luescher et al., 1991; Luescher, 2003). Obsessive compulsive behaviours in people include repetitive behaviours, such as hand washing, rituals, checking, arranging/ordering, counting and hoarding, and are accompanied by intrusive thoughts, such as: concern of contamination; concern for symmetry; fear of harm; aggressive, religious or sexual thoughts; or pathologic doubt. Interestingly, the intrusive thoughts (obsessions) and the associated behaviours (compulsions) do not necessarily correspond.
The extent of the similarities between the human and canine conditions is not yet known. One similarity is that, overall, the behaviours in companion animals are amenable to the same pharmacologic treatment as are obsessions and compulsions in people. However, there are differences between the human and canine conditions.
Presenting signs of compulsive disorder
The behaviours performed by dogs and cats with CD could be categorized as locomotory, oral, aggressive, vocalization and hallucinatory behaviours.
Cats that avoid imaginary objects, stare at shadows, or startle without obvious cause may fall into the same category.
Causes of compulsive disorder
Stress
Compulsive behaviours are considered to be conflict behaviours caused by environmentally induced conflict, frustration or stress. Therefore, any environmental factor resulting in frustration (e.g. lack of exercise), conflict
(e.g. inconsistent interaction) or stress (e.g. dominance conflict with another dog or separation anxiety) may contribute to the CD.
Genotype
A genetic predisposition may be present in any case of CD. Individuals may be genetically susceptible to development of a compulsive behaviour, or the genotype may determine which, if any, compulsive behaviour an animal develops. Apparent breed predispositions include flank sucking in Dobermans; spinning or freezing with the head under or between objects, such as clothes, in bull terriers; tail chasing in German shepherds and Australian cattle dogs (Luescher, 2003); and checking the hind end in miniature schnauzers. Furthermore, large-breed dogs seem to be more likely than small-breed dogs to develop lick granulomas. In cats, it seems that Siamese and Burmese breeds are particularly prone to develop skin sucking (Luescher et al., 1991). Temperament traits, such as genetic fearfulness, are likely to contribute to the development of a CD as well.
Medical problems
In some cases, a dog may start to lick a lesion or skin incision but then also start to lick other parts of the body. Persistent licking may cause lick granulomas at sites unrelated to the original lesion. This suggests that the stress associated with physical lesions or irritations, such as those caused by allergy, can contribute to the development of CD in an already susceptible animal and that the irritation can initially direct the compulsive behaviour toward a particular body site. Any other disease that increases stress and/or irritability, such as dermatologic disease or endocrine imbalance, may contribute to CD as well.
Conditioning
Most owners pay attention to their pets when they perform a compulsive behaviour. Therefore, most cases of CD may be aggravated by inadvertent conditioning. Performance of the behaviour only in the owner’s presence is suggestive of a purely conditioned behaviour.
Pathophysiology of compulsive disorder
The pathophysiology of CD is not well understood. Most evidence stems from drug effects on the performance of compulsive behaviour. Large doses of dopaminergic drugs, such as amphetamine or apomorphine, are effective in inducing stereotyped behaviour in animals, whereas the dopamine antagonist haloperidol results in suppression of spontaneously occurring stereotyped behaviour (Kennes et al., 1988). The role b-endorphins play in the development of compulsive behaviour is not known, but it has been suggested that they play a significant role only in the initial stages of stereotypy development (Hewson et al., 1998; Kennes et al., 1988). The b-endorphin antagonists can be effective in suppressing compulsive behaviours, but their application in clinical cases is not practical. Similar to the treatment of human OCD, drugs inhibiting serotonin reuptake have been found to be effective in the treatment of CD in dogs (Hewson et al., 1998). The effectiveness of such drugs implies that serotonin is involved in animal CD. Direct evidence of serotonin involvement has also been presented (Luescher, 2003).
Homogeneity of compulsive disorder
Aside from the fact that several of the behaviours may be found to have other possibly medical or neurologic reasons in the future, there is some evidence that CD is not a homogeneous condition and that there may be different classes of compulsive behaviour.
Development
The categories of locomotory and oral compulsive behaviours seem to differ. In general, locomotory compulsive behaviours start in one context and gradually generalize to other contexts in which the animal is agitated. Oral self-directed behaviours, however, seem to be displayed suddenly without identifiable initial conflict and are performed at a constant rate in contexts with little outside stimulation
(i.e. when the animal seems quiet (although its arousal level may be high) ). Owners often describe that it appears as if the dog had to perform the oral compulsive behaviour so as to be able to settle down. Neurophysiologic studies also seem to justify this categorization. It was suggested that oral stereotypic behaviours may involve the mesolimbic dopaminergic system, whereas locomotory stereotypic behaviours may involve activation of the nigrostriatal dopaminergic system.
Level of cognition involved
Some behaviours, such as fly biting or spinning, seem to involve little cognition and seem more akin to ‘tic’ disorders in human beings. Other CDs, however, seem to involve a high level of cognition. For example, miniature schnauzers that are hind-end checkers are not simply looking at their hind end in a repetitive constant fashion. First, they may turn either way. More importantly, they may get up and check the floor where they have been sitting and perhaps even scratch it. This implies that they actually perceive that there is something wrong. Dogs that chase light reflections may wait in the morning in the appropriate location where they know the rising sun produces the first light reflections. Dogs that are fixated on a particular object may look for that object when it is removed.
Ease of distraction
Some patients can easily be distracted with an innocuous noise, such as clicking of the tongue.
In other cases, the behaviour can only be interrupted by physically interfering with the behaviour (e.g. by pulling on a leash attached to a head halter). In a clinical trial investigating the efficacy of clomipramine in the treatment of CD, there was no difference in response to drug treatment between locomotory and oral compulsive behaviours (Hewson et al., 1998).
Clinical approach to compulsive disorder
Diagnosis
A diagnosis of CD is primarily based on a detailed history and on ruling out other possible causes for the observed behaviour. The history includes information on the life history
To exclude other possible causes for the behaviour, a minimal database consisting of a complete blood cell count (CBC), chemistry profile and urinalysis should be obtained. Additionally a thorough neurologic examination should determine if neurologic problems are present or not. If the results are normal, further neurologic testing may not be warranted but structural disease affecting the ‘silent’ areas of the brain should be considered, which would require advanced imaging such as MRI. An EEG during the event or at least in between the events should be considered as occasionally this can help determine that these events are seizure manifestations. Circling bull terriers were reported to have an abnormal EEG indicative of seizure activity (Dodman et al., 1996).
Compulsive behaviours are always displayed outside their natural context, usually in several different contexts, and/or are excessive. They are often directed toward unusual target objects and are frequently repetitive or sustained. The animal is fully conscious while performing the behaviour and aware of its surroundings (although it may not respond to any stimuli in the environment in some cases and may even run into furniture, for example). The behaviour can usually be interrupted, and the animal does not exhibit a post-ictal phase characteristic of seizures. Performance of the behaviour is not dependent on the owner’s presence (to exclude attention-getting behaviours). Locomotory compulsive behaviour and fly snapping are typically initially shown in a specific conflict situation and, later, in an increasing number of situations in which the animal is excited. Self-directed oral compulsive behaviours are likely to be shown in situations with little external stimulation.
Animals performing compulsive behaviours are aware of their surroundings, can usually be distracted (although sometimes with difficulty) from performing their behaviour, and, most importantly, do not show a post-ictal phase. As opposed to seizures, animals perform the compulsive behaviour usually when alert and interacting with their environment.
Treatment options
Because a CD derives from conflict behaviour, an attempt should be made to identify and remove the cause of conflict, frustration and stress. In cases where the cause of stress cannot be removed, it may be possible to desensitize the animal to the stressful situation. Lack of predictability and control over the environment is an important stress-inducing factor and may arise from inconsistent owner– animal interaction, lack of training to commands and thus inconsistent use of commands.
The inappropriate use of punishment, an inconsistent routine and frustration of motivations, such as the motivation for social interaction, are additional contributing factors.
Casual interaction should therefore be avoided and replaced with highly structured interactions in a command-response-reward format. Formal obedience sessions allow for such consistent interaction with dogs and establish a habit of using consistent commands in everyday situations.
If the animal engages in an inappropriate behaviour, it is distracted with a noise, a command is issued, and the animal is rewarded for obeying the command. A regular routine increases the predictability of the animal’s environment. It is particularly important that feeding and exercise become a consistent daily part of the owner’s routine. Sufficient exercise provided to dogs serves to fill their need for exploration and social interaction with other dogs, even if just by sniffing and leaving scent marks. Rotating toys maintains the animal’s interest in them and may provide an opportunity to reduce arousal. Particularly attractive toys, such as food-dispensing toys, can be given at times when the performance of the compulsive behaviour is likely.
In most cases, drug therapy may prove necessary or at least facilitate treatment. Pharmacologic intervention is most likely achieved with serotonin reuptake inhibitors, although it may take 4 weeks or longer to see an effect. A clinical trial involving 51 dogs with a variety of compulsive behaviours has proven the effectiveness of the tricyclic antidepressant clomipramine (Hewson et al., 1998).
A case series suggested effectiveness of clomipramine for tail chasing in terriers (Moon-Fanelli and Dodman, 1998). Clinical trials on cases of acral lick dermatitis have been performed for clomipramine, fluoxetine and sertraline (Rapoport et al., 1992). Paroxetine has also been used clinically, but its effect has not been evaluated. The drug is given until at least 3 weeks after it seems to have had a satisfactory effect and then weaned off gradually over at least 3 weeks by reducing dose but maintaining dosing frequency. If the behaviour reappears during the weaning process, the dose is increased again and maintained at the effective level for some time before resuming weaning. Weaning is important so as to avoid a rebound effect. Clomipramine can be combined with fluoxetine to slow down its metabolism. Tricyclics other than clomipramine or other anxiolytic drugs, such as benzodiazepines or buspirone, are unlikely to have any effect on CD on their own, and their use in drug combination therapy has not been evaluated in dogs. b-endorphin antagonists have been used experimentally, but most are injectables and have a short half-life, and their use for clinical cases is not practical (Garrett and el-Koussi, 1985; Dodman et al., 1988). The dopamine antagonist haloperidol has also not been proven effective in practice, most likely because an appropriate dosing regimen has not been established.
The main goal of drug treatment is to reduce the frequency of the compulsive behaviour to the point that behavioural modification
(i.e. response substitution) becomes practical. In dogs, the patient is initially trained with positive reinforcement to perform a desirable behaviour that is incompatible with (i.e. cannot be performed at the same time as) the compulsive behaviour. Whenever the dog cannot be supervised, it is put into a situation where it cannot perform the compulsive behaviour (e.g. the dog can be crated if it does not perform the behaviour in the crate). Every time the dog shows any inclination to perform the compulsive behaviour, it is distracted (if necessary, by pulling on a leash connected to a head halter). The command for the alternate behaviour is then given. The dog either performs or is made to perform the alternate behaviour and is then rewarded. The reward can be progressively delayed so that the dog has to stay in the chosen position for increasingly longer times before the reward is given. Some serious cases of CD have been treated successfully with this behavioural modification technique alone (i.e. without the use of drugs) (Luescher, 2003).
The distraction is important. If the dog is not distracted before a command (i.e. attention) is given, the treatment attempt could result in aggravation of the problem through inadvertent reinforcement of the behaviour. In cats, a similar programme is recommended. The cat is continuously supervised or placed in a position in which it does not perform the behaviour. Every time the cat is about to perform the compulsive behaviour, it is distracted (startled) and its attention is then reoriented by throwing a toy. Cats can also be clicker-trained and then treated as described for dogs (Luescher, 2003).
Prognosis
Analysis of a referral case-load showed that approximately two-thirds of cases improved to the client’s satisfaction and that outcome was negatively affected by problem duration (Luescher, 2003). It is therefore important to treat CD as early as possible.
References
Arthur, W. and Kaye, G.C. (2001) Current investigations used to assess syncope. Postgraduate Medical Journal 77, 20–3.
Bhatti, S.F., Vanhaesebrouck, A.E., Van Soens, I., Martle, V.A., Polis, I.E., Rusbridge, C. and Van Ham, L.M. (2011) Myokymia and neuromyotonia in 37 Jack Russell terriers. Veterinary Journal 189, 284–288.
Boeve, B.F., Dickson, D.W., Olson, E.J., Shepard, J.W., Silber, M.H., Ferman, T.J., Ahlskog, J.E. and Benarroch, E.E. (2007a) Insights into REM sleep behavior disorder pathophysiology in brainstem-predominant Lewy body disease. Sleep Medicine 8, 60–64.
Boeve, B.F., Silber, M.H., Saper, C.B., Ferman, T.J., Dickson, D.W., Parisi, J.E., Benarroch, E.E., Ahlskog, J.E., Smith, G.E., Caselli, R.C., Tippman-Peikert, M., Olson, E.J., Lin, S.C., Young, T., Wszolek, Z., Schenck, C.H., Mahowald, M.W., Castillo, P.R., Del Tredici, K. and Braak, H. (2007b) Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 130, 2770–2788.
Bright, J.M. and Cali, J.V. (2000) Clinical usefulness of cardiac event recording in dogs and cats examined because of syncope, episodic collapse, or intermittent weakness: 60 cases (1997–1999). Journal of the American Veterinary Medical Association 216, 1110–1114.
Brisbare-Roch, C., Dingemanse, J., Koberstein, R., Hoever, P., Aissaoui, H., Flores, S., Mueller, C., Nayler, O., Van Gerven, J., De Haas, S.L., Hess, P., Qiu, C., Buchmann, S., Scherz, M., Weller, T., Fischli, W., Clozel, M. and Jenck, F. (2007) Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nature Medicine 13, 150–155.
Bush, W.W., Barr, C.S., Stecker, M.M., Overall, K.L., Bernier, N.M., Darrin, E.W. and Morrison, A.R. (2004) Diagnosis of rapid eye movement sleep disorder with electroencephalography and treatment with tricyclic antidepressants in a dog. Journal of the American Animal Hospital Association 40, 495–500.
Calvert, C.A., Jacobs, G.J. and Pickus, C.W. (1996) Bradycardia-associated episodic weakness, syncope, and aborted sudden death in cardiomyopathic Doberman Pinschers. Journal of Veterinary Internal Medicine 10, 88–93.
Calvert, C.A., Jacobs, G., Pickus, C.W. and Smith, D.D. (2000) Results of ambulatory electrocardiography in overtly healthy Doberman Pinschers with echocardiographic abnormalities. Journal of the American Veterinary Medical Association 217, 1328–1332.
Dauvilliers, Y., Arnulf, I. and Mignot, E. (2007) Narcolepsy with cataplexy. Lancet 369, 499–511.
Delucchi, L., Martino, P., Baldovino, A., Pessina, P. and Rodrigues, A. (2010) Use of venlafaxine in the treatment of a canine narcolepsy-cataplexy case. Journal of Small Animal Practice 51, 132.
Dodman, N.H., Shuster, L., White, S.D., Court, M.H., Parker, D. and Dixon, R. (1988) Use of narcotic antagonists to modify stereotypic self-licking, self-chewing, and scratching behavior in dogs Journal of the American Veterinary Medical Association 193, 815–819.
Dodman, N.H., Knowles, K.E., Shuster, L., Moon-Fanelli, A.A., Tidwell, A.S. and Keen, C.L. (1996) Behavioral changes associated with suspected complex partial seizures in bull terriers. Journal of the American Veterinary Medical Association 208, 688–691.
Fujiki, N., Yoshida, Y., Ripley, B., Mignot, E. and Nishino, S. (2003) Effects of IV and ICV hypocretin-1 (orexin A) in hypocretin receptor-2 gene mutated narcoleptic dogs and IV hypocretin-1 replacement therapy in a hypocretin-ligand-deficient narcoleptic dog. Sleep 26, 953–959.
Galano, H.R., Olby, N.J., Howard, J.F., Jr and Shelton, G.D. (2005) Myokymia and neuromyotonia in a cat. Journal of the American Veterinary Medical Association 227, 1608–1612, 1591.
Garosi, L.S., Rossmeisl, J.H., De Lahunta, A., Shelton, G.D. and Lennox, G. (2005) Primary orthostatic tremor in Great Danes. Journal of Veterinary Internal Medicine 19, 606–609.
Garrett, E.R. and El-Koussi, A.E.-D. (1985) Pharmacokinetics of morphine and its surrogates V: Naltrexone and naltrexone conjugate pharmacokinetics in the dog as a function of dose. Journal of Pharmacological Science 74, 50–56.
Gill, J.L., Capper, D., Vanbellinghen, J.F., Chung, S.K., Higgins, R.J., Rees, M.I., Shelton, G.D. and Harvey, R.J. (2011) Startle disease in Irish wolfhounds associated with a microdeletion in the glycine transporter GlyT2 gene. Neurobiology of Disease 43, 184–189.
Gill, J.L., Tsai, K.L., Krey, C., Noorai, R.E., Vanbellinghen, J.F., Garosi, L.S., Shelton, G.D., Clark, L.A. and Harvey, R.J. (2012) A canine BCAN microdeletion associated with episodic falling syndrome. Neurobiology of Disease 45, 130–136.
Goodwin, J.K. (1998) Holter monitoring and cardiac event recording. Veterinary Clinics of North America - Small Animal Practice 28, 1391–1407, viii.
Grubb, B.P. (1999) Pathophysiology and differential diagnosis of neurocardiogenic syncope. American Journal of Cardiology 84, 3Q–9Q.
Hewson, C.J., Luescher, U.A., Parent, J.M., Conlon, P.D. and Ball, R.O. (1998) Efficacy of clomipramine in the treatment of canine compulsive disorder. Journal of the American Veterinary Medical Association 213, 1760–1766.
Jankovic, J. and Fahn, S. (1980) Physiologic and pathologic tremors. Diagnosis, mechanism, and management. Annals of Internal Medicine 93, 460–465.
John, J., Wu, M.F., Maidment, N.T., Lam, H.A., Boehmer, L.N., Patton, M. and Siegel, J.M. (2004) Developmental changes in CSF hypocretin-1 (orexin-A) levels in normal and genetically narcoleptic Doberman pinschers. Journal of Physiology 560, 587–592.
Johnson, L., Boon, J. and Orton, E.C. (1999) Clinical characteristics of 53 dogs with Doppler-derived evidence of pulmonary hypertension: 1992-1996. Journal of Veterinary Internal Medicine 13, 440–447.
Johnsrude, C.L. (2000) Current approach to pediatric syncope. Pediatric Cardiology 21, 522–531.
Kennes, D., Odberg, F.O., Bouquet, Y. and De Rycke, P.H. (1988) Changes in naloxone and haloperidol effects during the development of captivity-induced jumping stereotypy in bank voles. European Journal of Pharmacology 153, 19–24.
Kroeger, D. and De Lecea, L. (2009) The hypocretins and their role in narcolepsy. CNS & Neurological Disorders Drug Targets 8, 271–280.
Kube, S.A., Vernau, K.M. and Lecouteur, R.A. (2006) Dyskinesia associated with oral phenobarbital administration in a dog. Journal of Veterinary Internal Medicine 20, 1238–1240.
Lindberg, J., Saetre, P., Nishino, S., Mignot, E. and Jazin, E. (2007) Reduced expression of TAC1, PENK and SOCS2 in Hcrtr-2 mutated narcoleptic dog brain. BMC Neuroscience 8, 34.
Luescher, A.U. (2003) Diagnosis and management of compulsive disorders in dogs and cats. Veterinary Clinics of North America - Small Animal Practice 33, 253–267, vi.
Luescher, U.A., Mckeown, D.B. and Halip, J. (1991) Stereotypic or obsessive-compulsive disorders in dogs and cats. Veterinary Clinics of North America - Small Animal Practice 21, 401–413.
Meurs, K.M., Spier, A.W., Wright, N.A. and Hamlin, R.L. (2001) Use of ambulatory electrocardiography for detection of ventricular premature complexes in healthy dogs. Journal of the American Veterinary Medical Association 218, 1291–1292.
Mignot, E. (2004) An update on the pharmacotherapy of excessive daytime sleepiness and cataplexy. Sleep Medicine Reviews 8, 333–338.
Miller, R.H., Lehmkuhl, L.B., Bonagura, J.D. and Beall, M.J. (1999) Retrospective analysis of the clinical utility of ambulatory electrocardiographic (Holter) recordings in syncopal dogs: 44 cases (1991-1995). Journal of Veterinary Internal Medicine 13, 111–122.
Mishima, K., Fujiki, N., Yoshida, Y., Sakurai, T., Honda, M., Mignot, E. and Nishino, S. (2008) Hypocretin receptor expression in canine and murine narcolepsy models and in hypocretin-ligand deficient human narcolepsy. Sleep 31, 1119–1126.
Moon-Fanelli, A.A. and Dodman, N.H. (1998) Description and development of compulsive tail chasing in terriers and response to clomipramine treatment. Journal of the American Veterinary Medical Association 212, 1252–1257.
Nishino, S. (2007) Clinical and neurobiological aspects of narcolepsy. Sleep Medicine 8, 373–399.
Packer, R.A., Patterson, E.E., Taylor, J.F., Coates, J.R., Schnabel, R.D. and O’Brien, D.P. (2010) Characterization and mode of inheritance of a paroxysmal dyskinesia in Chinook dogs. Journal of Veterinary Internal Medicine 24, 1305–1313.
Penderis, J. and Franklin, R.J. (2001) Dyskinesia in an adult bichon frise. Journal of Small Animal Practice 42, 24–25.
Platt, S.R., De Stefani, A. and Wieczorek, L. (2006) Primary orthostatic tremor in a Scottish deerhound. Veterinary Record 159, 495–496.
Podell, M. (2004) Tremor, fasciculations, and movement disorders. Veterinary Clinics of North America - Small Animal Practice 34, 1435–1452.
Ramsey, I.K., Chandler, K.E. and Franklin, R.J. (1999) A movement disorder in boxer pups. Veterinary Record 144, 179–180.
Rapoport, J.L., Ryland, D.H. and Kriete, M. (1992) Drug treatment of canine acral lick. An animal model of obsessive-compulsive disorder. Archives of General Psychiatry 49, 517–521.
Schatzberg, S.J., Cutter-Schatzberg, K., Nydam, D., Barrett, J., Penn, R., Flanders, J., Delahunta, A., Lin, L. and Mignot, E. (2004) The effect of hypocretin replacement therapy in a 3-year-old Weimaraner with narcolepsy. Journal of Veterinary Internal Medicine 18, 586–588.
Schnipper, J.L. and Kapoor, W.N. (2001) Diagnostic evaluation and management of patients with syncope. The Medical Clinics of North America 85, 423–56, xi.
Schubert, T.A., Chidester, R.M. and Chrisman, C.L. (2011) Clinical characteristics, management and long-term outcome of suspected rapid eye movement sleep behaviour disorder in 14 dogs. Journal of Small Animal Practice 52, 93–100.
Shelton, G.D. (2004) Muscle pain, cramps and hypertonicity. Veterinary Clinics of North America - Small Animal Practice 34, 1483–1496.
Smedile, L.E., Duke, T. and Taylor, S.M. (1996) Excitatory movements in a dog following propofol anesthesia. Journal of the American Animal Hospital Association 32, 365–368.
Tonokura, M., Fujita, K., Morozumi, M., Yoshida, Y., Kanbayashi, T. and Nishino, S. (2003) Narcolepsy in a hypocretin/orexin-deficient chihuahua. Veterinary Record 152, 776–779.
Tonokura, M., Fujita, K. and Nishino, S. (2007) Review of pathophysiology and clinical management of narcolepsy in dogs. Veterinary Record 161, 375–380.
Van Ham, L., Bhatti, S., Polis, I., Fatzer, R., Braund, K. and Thoonen, H. (2004) ‘Continuous muscle fibre activity’ in six dogs with episodic myokymia, stiffness and collapse. Veterinary Record 155, 769–774.
Vanhaesebrouck, A.E., Bhatti, S.F., Bavegems, V., Gielen, I.M., Van Soens, I., Vercauteren, G., Polis, I. and Van Ham, L.M. (2010a) Inspiratory stridor secondary to palatolingual myokymia in a Maltese dog. Journal of Small Animal Practice 51, 173–175.
Vanhaesebrouck, A.E., Van Soens, I., Poncelet, L., Duchateau, L., Bhatti, S., Polis, I., Diels, S. and Van Ham, L. (2010b) Clinical and electrophysiological characterization of myokymia and neuromyotonia in Jack Russell Terriers. Journal of Veterinary Internal Medicine 24, 882–889.
Vanhaesebrouck, A.E., Shelton, G.D., Garosi, L., Harcourt-Brown, T.R., Couturier, J., Behr, S., Harvey, R.J., Jeffery, N.D., Matiasek, K., Blakemore, W.F. and Granger, N. (2011) A novel movement disorder in related male Labrador Retrievers characterized by extreme generalized muscular stiffness. Journal of Veterinary Internal Medicine 25, 1089–1096.
Wagner, S.O., Podell, M. and Fenner, W.R. (1997) Generalized tremors in dogs: 24 cases (1984-1995). Journal of the American Veterinary Medical Association 211, 731–735.
Walmsley, G.L., Smith, P.M., Herrtage, M.E. and Jeffery, N.D. (2006) Facial myokymia in a puppy. Veterinary Record 158, 411–412.
Ware, W.A. (1999) Twenty-four-hour ambulatory electrocardiography in normal cats. Journal of Veterinary Internal Medicine 13, 175–180.
Ware, W.A. and Hopper, D.L. (1999) Cardiac tumors in dogs: 1982-1995. Journal of Veterinary Internal Medicine 13, 95–103.
Willis, R., Mcleod, K., Cusack, J. and Wotton, P. (2003) Use of an implantable loop recorder to investigate syncope in a cat. Journal of Small Animal Practice 44, 181–183.
Wolf, M., Bruehschwein, A., Sauter-Louis, C., Sewell, A.C. and Fischer, A. (2011) An inherited episodic head tremor syndrome in Doberman pinscher dogs. Movement Disorders 26, 2381–2386.
Wu, M.F., John, J., Maidment, N., Lam, H.A. and Siegel, J.M. (2002) Hypocretin release in normal and narcoleptic dogs after food and sleep deprivation, eating, and movement. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology 283, R1079–1086.
Wu, M.F., Nienhuis, R., Maidment, N., Lam, H.A. and Siegel, J.M. (2011) Role of the hypocretin (orexin) receptor 2 (Hcrt-r2) in the regulation of hypocretin level and cataplexy. Journal of Neuroscience 31, 6305–6310.
10 Clinical and Diagnostic Investigation of the Seizure Patient
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
A seizure is ‘a transient occurrence of signs due to abnormal excessive or synchronous neuronal activity in the brain’ (Fisher et al., 2005), which may manifest in different ways and be caused by various underlying aetiologies (see Chapter 3, Tables 3.2, 3.3 and Boxes
3.1 and 3.2). The clinical manifestations of various disorders such as narcolepsy/cataplexy or other sleep disorder, neuromuscular collapse, metabolic collapse (e.g. hypoglycaemia), movement disorder (dyskinesia), syncope, and vestibular event may be misinterpreted as seizures (see Chapter 9, Table 9.1). When investigating animals presenting with a history of seizures the fundamental questions to answer are:
The answer to the first question can be quite simple in animals with generalized tonicclonic seizures with loss of consciousness and autonomic signs (e.g. hypersalivation, urination and defecation). However, other clinical manifestations (e.g. focal seizure or atonic seizure) may be more challenging to classify as seizure versus another disorder. Ultimately the epileptic nature of an event can be confirmed only with simultaneous observation of electroencephalographic (EEG) abnormalities and clinical manifestation of the seizures, however this is rarely feasible in veterinary patients. Most commonly the clinician relies on the pet owner’s detailed description and video footage of the event. The presence of pre-or post-ictal signs, the peracute and unpredictable onset and termination of the event, the presence of involuntary motor activity and/or abnormal mentation and behaviour and/or autonomic signs, and the stereotypical pattern of each event are all suggestive of seizure activity.
Based on the information obtained from signalment, a detailed history, physical and neurological examination, the clinician can formulate a list of differential aetiologic diagnoses and subsequently select and interpret diagnostic tests. The aetiologic diagnosis enables the initiation of specific treatment and predicting prognosis.
Signalment
Information on species, breed, gender and age (particularly age at seizure onset) may raise the degree of suspicion of certain seizure aetiologies. Dogs of certain breeds (see Chapter 6, Table 6.1) with seizure onset between 6 months and 6 years of age and no interictal clinical abnormalities are likely to have idiopathic epilepsy. Congenital or developmental diseases occur most often in young animals, whereas neoplasia is more common in older patients. Puerperal hypocalcaemia would occur only in nursing female dogs (Thomas, 2010).
Medical History
A detailed and accurate history is the foundation of investigation of the seizure patient. Obtaining a good history requires time, patience, skill and experience. Using a vocabulary understandable to the pet owner, whilst showing compassion and empathy, allows the establishment of a positive relationship with the pet owner, which will assist in obtaining the most helpful information. The pet owner should feel comfortable in answering ‘I do not know’ or ‘I am not sure’ as this information is more valuable than misleading defensive answers (Thomas, 2010). In addition to the general questions that are routinely asked while taking the history of any neurologic patient (see Box 10.1) specific additional questions should be asked to ascertain the presence and clinical characteristics of seizure
Box 10.1. General information required while taking the history of the neurologic patient.
activity as well as any other neurological or behavioural abnormalities (see Box 10.2). In animals with interictal neurological abnormalities, information on onset (peracute, acute, subacute, chronic), course (static, progressive, relapsing remitting, regressive) and any factors (including treatment) affecting disease course help to formulate the differential aetiologic diagnosis list (see Table 10.3).
General Physical Examination
A thorough general physical examination can detect signs of a systemic or other organ illness that could be the underlying cause of the seizures or may affect seizure management
(e.g. concurrent renal disease will affect selection and dosage of anti-epileptic medications (AEM)). Worn nails may be the result of chronic proprioceptive deficits.
Neurological Examination
The aims of the neurological examinations are
to investigate if the nervous system is affected and if so, to identify location (neuroanatomic diagnosis) and distribution (e.g. focal, multi-focal or diffuse, lateralized or symmetrical) of the lesion. Seizure activity already indicates involvement of the forebrain. The presence of other neurologic signs and the resulting neuroanatomic diagnosis will affect the differential aetiologic diagnosis list and subsequently selection and interpretation of diagnostic investigations.
The neurological examination tests the functional integrity of the various components of the nervous system and can be subdivided into evaluation of level of consciousness and behaviour, posture, gait, cranial nerve function, postural reactions, muscle mass and tone, spinal nerve reflexes, nociception, and palpation of the head, spine and muscles. The order in which these components are evaluated can change depending on the individual patient and clinician preference. Parts of the neurological examination can be integrated with the general physical examination. The neurologic examination findings should be noted in a specific form (Box 10.3).
Box 10.2. Specific information required when animals present for seizure investigation.
Age at seizure onset Number, frequency, pattern and if available, date and time of the day of seizures Description of what the pet was doing prior to the seizure/s (sleeping, resting, walking, running, eating, fasting) Presence of an apparent triggering factor (e.g. stressful events, feeding, exercise) Description and duration of any behavioural abnormalities observed immediately prior to seizure onset (prodrome) (e.g. anxiety, restlessness, increased affection, withdrawal, hiding, aggressiveness or vocalization) Detailed description of the seizure (ictus) (review video footage anytime possible) (e.g. motor and autonomic manifestations, alteration of consciousness/awareness, stereotypical movements) Duration of ictus (ideally based on assessment with a timing device) Description and duration of any abnormalities observed immediately after the seizure (post-ictal phase)
(e.g. disorientation, aggressiveness, restlessness, pacing, ataxia, lethargy, deep sleep, excessive hunger or thirst, blindness) Consistency or variability of clinical manifestations during the prodrome, aura (if recognized), ictus and post-ictal period Possibility to interrupt the episode by distracting or stimulating the animal Presence, onset and course of any interictal abnormalities such as behavioural changes (e.g. loss of house training, inability to follow commands, increased affection, isolation or aggression, disruptions in the pet’s normal sleep-wake cycle) or neurological deficits (e.g. altered mental status, aimless pacing, a tendency to turn and circle to one side, head pressing, apparent blindness) Administered anti-epileptic medications (AEMs) (type, dosage, serum levels and associated clinical response)
The following description of the neurological examination is done with an emphasis on evaluation of animals presenting with a history of seizures. Other textbooks (De Lahunta and Glass, 2009; Lorenz et al., 2011; Garosi and Lowrie, 2013a) should be consulted for further information of functional neuroanatomy or details of neurological findings in animals with extracerebral neurological disorders.
During the neurological examination, the clinician should make any effort to promote the animal’s cooperation. Initially, the animal should be allowed to explore the consultation room while the history is taken. The clinician can observe the patient moving freely and assess mental status, behaviour, posture, gait and presence of involuntary movement abnormalities. In the meantime, the animal can familiarize with the new environment and be more relaxed during the ‘hands on’ examination. Animals that present with a history of trauma potentially resulting in traumatic brain as well as spinal injury should be immobilized until vertebral instability has been ruled out and undergo a partial assessment prioritizing organ systems and injuries based on an emergency triage system.
Level of consciousness and behaviour
Level of consciousness and behaviour are assessed by observing the animal’s response to the environment and attitude to being handled by its owner and the clinician. Responses to various environmental stimuli should be interpreted in the context of normality for species, breed and age. Information obtained from the pet owner can help to establish whether the responses of the individual should be interpreted as normal or abnormal. Dogs should be observed in the consultation room and, whenever possible, also outside the clinic where they are more likely to be relaxed and environmental stimulation may be richer (Fig. 10.1). Cats may be placed on a window sill in order to observe their response to the outside environment, especially if other animals are visible (Fig. 10.2).
Level of consciousness (awareness, mental state) relies on the functional integrity of the ascending reticular activating system (ARAS) within the brainstem and of the cerebral cortex (Plate 23). The ARAS receives sensory stimuli from inside and outside the body and through the ARAS




diencephalic portion projects diffusely to the cerebral cortex. Animals with normal level of consciousness are alert and respond appropriately to the environment. Obtunded animals are relatively inactive and unresponsive to the environment. They tend to sleep when undisturbed and arouse following tactile or acoustic stimuli. Obtundation may be caused by systemic disease (e.g. fever, anaemia) as well as by diffuse cerebral cortical disease or a brainstem lesion. Stuporous (semicomatose) animals tend to sleep when undisturbed and vigorous tactile stimuli, loud noise or noxious stimuli are necessary to arouse them. Comatose animals are unconscious and cannot be aroused even with noxious stimuli, although reflexes (e.g. palpebral, patellar, withdrawal) may remain intact. Stupor and coma indicate partial or complete disconnection between the ARAS and the cerebral cortex, respectively (Lorenz et al., 2011). Delirium is characterized by hyperactivity and exaggerated responses to environmental stimuli (Thomas, 2010). Disoriented animals appear confused and not fully aware of the environment and respond inappropriately to environmental stimuli.
Behaviour is controlled by the limbic system, which consists of portions of the cerebrum and diencephalon. Behavioural changes and seizures may be the only clinical abnormalities in animals with metabolic and structural forebrain disorders. In addition, behavioural changes such as fear/anxiety, defensive aggression and abnormal perception (e.g. barking without apparent cause, chasing shadows or light spots, aimless pacing, staring into space) have been reported to occur with the development of idiopathic epilepsy in dogs (Shihab et al., 2011). Restlessness and sedation may also occur as adverse effects to phenobarbital and potassium bromide (Chang et al., 2006). Behavioural disorders may also result from changes in the animal’s environment, previous experience or training. Subtle changes in the pet’s behaviour can be detected only by the pet owner in the animal’s normal environment and this information should be obtained while taking the history. Behavioural abnormalities that can be observed during the consultation include: withdrawal, aggression, aimless pacing, compulsive walking, tendency to turn and circle to one side (circling) and head pressing (Fig. 10.3). Lateralized forebrain disorders may cause a peculiar neurologic abnormality known as hemi-neglect or hemiinattention syndrome. This is characterized by the animal’s lack of response to any environmental stimulus on one side of its body (which is generally contralateral to the affected forebrain). The owner may report that the animal eats from one side of the bowl only (Fig. 10.4).
Posture
The position of the animal’s head, neck, trunk, limbs and tail are carefully observed. Normal posture requires integrity of the proprioceptive (general and special) and motor systems as well as the cerebellum. Postural abnormalities associated with lateralized forebrain disease include head turn and pleurothotonus (Fig. 10.5a–c). Head turn is characterized by the head being turned to one side while its median plane remains perpendicular to the ground (the ears are level). Pleurothotonus refers to a head, neck and trunk turn to one side. Usually, but not always, the direction toward which the animal turns the head, neck and trunk and tends to walk compulsively in circles is ipsilateral to the affected forebrain (frontoparietal lobe or rostral thalamic) side.

Head tilt is characterized by a rotation of the median plane of the head so that one ear is lower than the other (Fig. 10.6). Head tilt generally results from vestibular or vestibulocerebellar (caudal cerebellar peduncle, flocculonodular lobe, or the fastigial nucleus) disease.
A low head carriage is a sign of neck pain generally associated with cervical disease (Fig. 10.7), however it may also occur with intracranial disease resulting in compression or stretching of the meninges or cerebral vasculature (Fig. 10.5). Animals with low head carriage associated with cervical disease such as meningitis, radiculitis, intervertebral disc extrusion, myositis, polyarthritis and vertebral fracture/luxation, tend to be reluctant to move the neck and often present with spasm of the cervical muscles.
Cervical ventroflexion, palmigrade and plantigrade stance are generally associated with neuromuscular weakness. Scoliosis, kyphosis and lordosis indicate lateral, dorsal and ventral deviation of the vertebral column, respectively. Torticollis indicates twisting of

Fig. 10.4. Hemi-neglect syndrome in an 8-year-old weimaraner who underwent right-sided transfrontal craniectomy to remove a right olfactory bulb meningioma. The dog had been eating from the right side of the bowl only since before the surgery. It was initially presented for cluster of focal seizures with secondary generalization.
the neck. These postures are generally associated with spinal diseases. A wide-based stance may occur with general proprioceptive, vestibular or cerebellar disorders and sometimes also with generalized weakness.
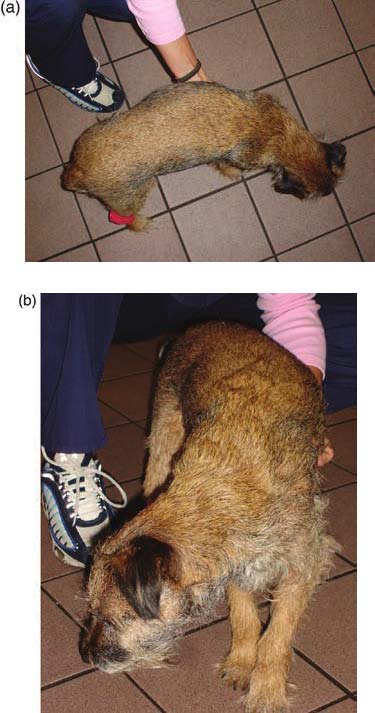
Fig. 10.5 Pleurothotonus (head, neck and trunk turn) to the right, low head carriage and decreased level of consciousness in a 7-year 9-month-old, female spayed, Border terrier with a 2-week history of circling to the right and obtundation (a, b). MRI of the brain revealed extensive, irregular, poorly marginated areas of increased signal intensity on T2W images throughout the brain and particularly within the white matter of the right frontal lobe (c), temporal and right occipital lobes, as well as the thalamus and brainstem. Post-contrast T1W images showed meningeal enhancement but no enhancement of the brain parenchyma. CSF analysis revealed a severe lymphocytic pleocytosis.


Spontaneous knuckling of the toes generally indicates general proprioceptive dysfunction (Fig. 10.8).
Abnormal postures in recumbent animals include decerebrate rigidity, decerebellate rigidity and the Schiff-Sherrington posture. Decerebrate rigidity is characterized by extension of all limbs and opisthotonus (dorsiflexion of the head and neck) (Fig. 10.9). This posture is caused by a lesion in the rostral brainstem (midbrain), and affected animals typically have decreased consciousness (generally stupor or coma). Decerebellate rigidity occurs with acute lesions involving the rostral cerebellum, and is characterized by opisthotonus, thoracic limb extension and flexion of the hips. Consciousness is normal (Fig. 10.10). The Schiff-Sherrington posture can be observed in animals with acute and severe thoracolumbar spinal cord injuries and consists of increased extensor tone in the thoracic limbs and paralysis of the pelvic limbs (Fig. 10.11). This posture is caused by sudden loss of inhibitory input from the border cells (which are located in the L1 to L5 spinal cord segments) to the extensor motor neurons in the cervical intumescence.
Gait
The gait should be examined while the animal walks on a non-slippery surface. Dogs can be observed while moving freely in the consultation room as well as outside on a leash while walked and trotted in a straight line and in circles. Walking the dog up and down stairs can help to identify subtle deficits. Cats can be encouraged to walk around the room using toys they can chase or by moving their carrier in different areas of the room as they may want to hide back inside. If not hazardous, allowing cats to jump on and off a small height may help to identify subtle deficits. The animal should be observed from all directions, paying attention to movement coordination and strength. The clinician should be knowledgeable of differences in normal gait among species and breeds. Normal posture and gait require integrity of the proprioceptive (general and special) and motor systems as well as the cerebellum. Proprioception detects the position or movement of body parts. Receptors sensitive to movement and stretch are located in muscles, tendons and joints (general proprioception). This information is conveyed by peripheral nerves to the spinal cord, which integrates local reflexes involved in posture and movement. Proprioceptive information also travels through ascending spinal tracts to the brainstem, cerebellum and forebrain, which integrate coordinated movement. In addition, information on position and movement of the head are detected by the vestibular receptors (special proprioception) and conveyed to the vestibular nuclei in the brainstem, the cerebellum, the motor nuclei of CN III, IV and VI and to alpha-motor neurons of extensor and flexor muscles through the vestibulo-spinal tract. Conscious perception of balance is thought to be mediated by afferent pathways from the vestibular nuclei to the temporal cerebral cortex via thalamic relay centres. A lesion in the proprioceptive system or cerebellum results in ataxia and a lesion in the motor system results in paresis or paralysis. Ataxia refers to uncoordinated gait and can be subclassified as general proprioceptive, vestibular and cerebellar.


General proprioceptive ataxia is characterized by a loss of the sense of limb and body position. The limbs may be abducted and adducted excessively to the point they cross over during the protraction phase of the gait, and then over-flex during protraction. The digits are scuffed or dragged, and knuckled over during the support phase of the gait. The trunk may sway from side to side. General proprioceptive ataxia is caused by a lesion affecting the general proprioceptive pathways in the peripheral nerve, dorsal root, spinal cord, brainstem and forebrain. General proprioceptive pathways are anatomically close to motor pathways, and therefore general proprioceptive ataxia is often associated with paresis.
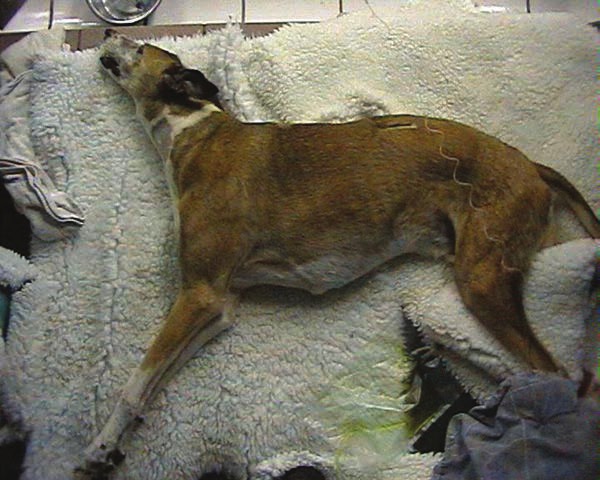

Vestibular ataxia associated with unilateral vestibular disease is characterized by a loss of balance resulting in a head tilt and a tendency to lean, drift, fall or roll to one side. Abnormal nystagmus and positional strabismus may also be present. Animals with bilateral vestibular dysfunction maintain a crouched position, are reluctant to move and have wide side-to-side head excursions. These animals have no vestibulo-ocular reflex and no abnormal nystagmus.
Cerebellar ataxia is caused by cerebellar disease and is characterized by dysmetria, which is the inability to regulate the rate and range of movement. This most commonly manifests as hypermetria, an over-reaching, high-stepping gait with marked over-flexion of the limbs on protraction. This is followed by over-extension of the limb during the support phase of the gait and a delay in the onset of protraction. The trunk may sway from side to side and a fine tremor of the head and neck may occur upon initiation of voluntary movements (intention tremor). The animal may have a wide-based stance. Involvement of the caudal cerebellar peduncle, flocculonodular lobe or the fastigial nucleus of the cerebellum may result in vestibular signs.

Paresis is defined as a deficiency in the generation of the gait or the ability to support weight (de Lahunta, 2009). The terms paresis and paralysis (or plegia) indicate a partial or complete loss of motor function, respectively. The prefix mono-, hemi-, para- and tetra- can be combined with paresis and plegia to indicate partial or complete loss of motor function in one limb, a thoracic and pelvic limb on the same side, both pelvic limbs and all four limbs, respectively. Motor dysfunction may result from upper motor neuron (UMN) (brain and spinal cord) or lower motor neuron (LMN) (spinal cord ventral horn, nerve root, spinal nerve, peripheral nerve, neuromuscular junction) lesions.
Lameness is usually due to pain resulting from orthopaedic disease or certain neurologic
(e.g. nerve root or spinal nerve) disorders. The stride of the affected limb is often shortened. If the limb is severely painful, the animal does not bear any weight on it. If multiple limbs are involved the animal may be reluctant to stand up and walk.
Circling refers to the animal’s tendency to walk in circles and may occur with unilateral vestibular or lateralized forebrain disease. Animals with lateralized forebrain diseases may present with head turn or pleurothotonus and propulsive circling to one side, which is generally ipsilateral to the forebrain lesion. The gait may appear otherwise normal (in animals with non-peracute disorders) and mentation and behaviour may be altered. Circling resulting from unilateral vestibular disease is associated with vestibular ataxia (impaired balance) and often also other vestibular signs
(e.g. head tilt, abnormal nystagmus and positional strabismus).
Involuntary movement abnormalities and cataplexy
The animal should be observed for abnormal involuntary movements or abnormal skeletal muscle activities while resting, standing up and moving. Seizure activity can result in various motor manifestations (see Chapter 3) and occasionally can occur during the clinical examination.
Myoclonus is the clinical manifestation of a sudden contraction, followed immediately by relaxation of a specific muscle group. It has been classified as sporadic or repetitive. Repetitive myoclonus has been subdivided into constant (e.g. with canine distemper virus encephalomyelitis), action-related (e.g. CNS dysmyelination) and postural (e.g. ‘head bobbing’). Action-related repetitive myoclonus diffusely affects skeletal muscle and is rapid, with many contractions and relaxations per second, producing what is commonly described as a tremor (de Lahunta and Glass, 2009).
Tremor is defined as alternating contractions of antagonistic muscle groups and may result from neurologic causes, primary muscle disease as well as fatigue, fear, chilling or drug reactions. Tremors that occur or worsen when an animal is trying to perform purposeful movements (intention tremors) are most often associated with cerebellar disease.
Myotonia is caused by a muscle cell membrane disorder and is characterized by sustained muscular contraction following a physiologic stimulus. Myotonia can be congenital (e.g. in the miniature schnauzer) or acquired (e.g. secondary to hyperadrenocorticism).
Myokymia refers to the undulating vermiform movement of the skin overlying rhythmically contracting myofibres. It has also been termed continuous muscle fibre activity, episodic repetitive myoclonus or neuromyokymia to indicate the role of lower motor neuron hyperexcitability in this disorder. Myokymia may be focal (localized to certain muscle groups) or generalized.
Neuromyotonia refers to sustained muscle contraction due to hyperexcitability of lower motor neurons, possibly due to abnormalities of the axonal voltage-gated potassium channels. Neuromyotonia is clinically characterized by persistent muscle contraction, muscle stiffness or cramps, and impaired muscle relaxation often resulting in rigidity of all four limbs leading to collapse in lateral recumbence. Neuromyotonic attacks are mostly triggered by excitement, exercise or hot weather and can last from a few minutes to several hours (Bhatti et al., 2011).
Tetanus is the clinical sign of persistent contraction of extensor muscles and is caused by disinhibition of extensor motor neurons. This is most commonly caused by the toxin produced by Clostridium tetani infection (de Lahunta and Glass, 2009).
Tetany is the clinical sign of variably intermittent contraction of extensor muscles. Strychnine intoxication causes tetany by interfering with the release of glycine from spinal cord interneurons (de Lahunta and Glass, 2009). Hypocalcaemia can also produce tetany as a result of neuronal and muscle hyperexcitability.
Dyskinesia (abnormal (‘dys’) movement (‘kinesia’)) is a general term, which encompasses various forms of sudden and involuntary contractions of muscle groups (movement disorders) either at rest or during activity in a conscious animal. Breed-specific paroxysmal dyskinesias have been described (see Chapter 9). Dyskinesia characterized by fine intermittent contractions of the facial muscles and ears, and severe intermittent contractions of the cervical and shoulder muscles that would cause the affected dog to fall, has been reported as a reversible adverse effect of phenobarbital treatment (Kube et al., 2006).
Consciousness remains normal in animals with dyskinesia, myoclonus, tremor, myotonia, myokymia, neuromyotonia, tetanus and tetany.
Cataplexy is the sudden onset of complete atonia (flaccid paralysis) of the skeletal muscles resulting in the collapse of the animal (or only a dropped jaw in less severe cases) without loss of consciousness. Cataplexy can be triggered by excitement (such as eating, playing or the presence of the owner or another dog), it lasts a few seconds to 20 min, it may occur multiple times a day and it is frequently associated with narcolepsy (a disorder of sleep/wake control) (see Chapter 9).
Cranial nerve (CN) examination
The animal’s head is observed to evaluate symmetry of the ear, lips and nostrils (CN VII), masticatory muscle bulk and symmetry (CN V), palpebral fissure size and symmetry (CN III, sympathetic supply), eye position and movements (CN III, IV, VI, VIII), and pupil size and symmetry (CN II, III-parasympathetic, sympathetic supply). Abnormalities of palpebral or pupil size and symmetry may be caused also by ocular disorders. Pupil size and symmetry can be evaluated using an indirect ophthalmoscope in a dimly lit room (Fig. 10.12). Loss of sympathetic supply to the eye results in miosis, upper and third eyelid ptosis, and enophthalmos (Horner’s syndrome) (Fig. 10.13). Vision (retina, CN II, optic chiasm, optic tract, lateral geniculate nucleus, optic radiation, visual cortex) (Plate 24) can be assessed by observing the animal’s movements in the consultation room, avoidance of obstacles and ability to follow moving objects. Visual following can be assessed by dropping cotton balls or moving a toy or ball (avoiding creating noise, air movement or mechanical vibrations) in front of the animal and observing whether the patient’s eyes and head follow the object (Fig. 10.14). The visual pathways are also assessed during the menace response, pupillary light reflex (PLR) and tactile placing response (see postural reactions). Blindness with a dilated, unresponsive


third eyelid ptosis, and enophthalmos) on the right eye in a cat with a traumatic injury to the right cervical sympathetic trunk.

pupil occurs with a lesion of the retina or optic nerve/chiasm/tract. Impaired vision with normal pupil size and PLR indicates a lesion of the forebrain at the level of the optic radiation or occipital cortex.
The menace response is elicited by making a threatening gesture with the hand at one eye while the opposite eye is covered with the other hand (Fig. 10.15). The normal response is eyelid closure. Animals unable to blink due to facial paralysis may show eyeball retraction (VI), third eyelid elevation or an aversive movement of the head. The menace response tests the integrity of the retina, CN II, contralateral central visual pathway, visual cortex and motor cortex, the ipsilateral cerebellar cortex and CN VII (Plate 25). The menace response is a learned response and it is generally present from 10 to 12 weeks of age in dogs and cats. In animals with forebrain disease, a decreased or absent menace response (uni- or bilaterally) may be the only or one of the few neurological abnormalities detected during the examination and therefore must be evaluated carefully in animals presenting with seizures.
The palpebral reflex is performed by touching the medial and lateral canthus of each eye, eliciting eyelid closure (CN V-ophthalmic (medial canthus) and maxillary (lateral canthus) branches; CN VII) (Fig. 10.16). In some animals, it helps to elicit the palpebral reflex a few times

Fig. 10.15. The menace response is elicited by making a threatening gesture at one eye, while the contralateral eye is blindfolded with the other hand. The normal response is closure of the eyelids. The examiner must avoid creating air currents or touching the hairs around the eye, which will elicit the palpebral reflex. The menace response pathway is illustrated in Fig. 10.18.
before testing the menace response as they are more likely to interpret the clinician’s hand gesture as threat.
The PLR is performed by shining a bright light source (e.g. Finoff trans-illuminator) in each eye, eliciting pupillary constriction in both eyes (retina, CN II, optic chiasm, optic tract, pretectal nuclei and CNIII (parasympathetic)) (Fig. 10.17, Plate 26). Iris abnormalities, such as iris atrophy, may result in decreased PLR. Abnormalities of pupil size and response to light can be observed in animals with cerebral oedema and herniation of the occipital lobe ventral to the tentorium, causing compression and displacement of the midbrain, oculomotor nerve (CN III) or both.

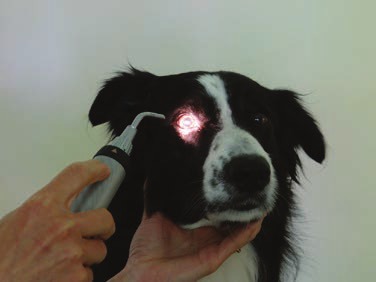
The vestibulo-ocular reflex (also named normal or physiologic nystagmus) (vestibular receptors in the membranous labyrinth, CN VIII-vestibular branch, vestibular nuclei and medial longitudinal fasciculus in the brain stem, CN III, IV, VI) is elicited by turning the animal’s head from side to side and up and down to observe conjugate eye movements related to movements of the head.
The presence of strabismus and abnormal nystagmus (CN III, IV, VI, VIII) should be evaluated with the animal’s head at rest and held in varying positions. Strabismus (abnormal position of the ocular globe) can be classified based on its direction as ventrolateral (CN III paralysis), medial (CN IV paralysis) and dorsolateral (or rotatory) (CN VI paralysis). Ventrolateral strabismus can be classified as static (CN III paralysis) when it is present in all head positions or positional (CN VIII paralysis) when it occurs only when the head position is changed (e.g. extended) (Fig. 10.18). Abnormal nystagmus can be classified based on the direction of the fast phase (right or left) and on the direction of the abnormal ocular globe movement (horizontal, vertical or rotatory). Resting abnormal nystagmus is observed when the head is in its normal position. Positional abnormal nystagmus occurs or changes in direction when the head position is changed. Bilateral strabismus and pendular nystagmus can occur with some congenital anomalies in the anatomy of the visual system central projections (e.g. Siamese cats).
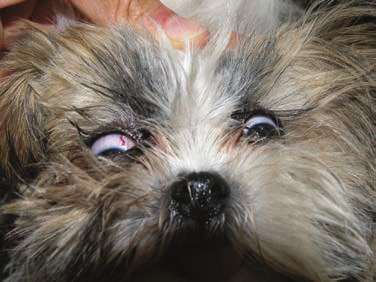
Facial sensation is evaluated by touching the areas innervated by the ophthalmic, maxillary and mandibular branches of CN V and observing a reflex (e.g. facial muscle twitch, CN VII) or a conscious response (e.g. head retraction) mediated at the level of the contra-lateral forebrain. The conscious response is best appreciated by covering both eyes and stimulating the nasal septal mucosa (CN V-ophthalmic) on each side with a haemostat (Fig. 10.19). Animals with forebrain disease may have facial hypo-algesia (contralateral to the affected forebrain) while palpebral and corneal reflexes remain normal.
The corneal reflex (CN V-ophthalmic, VI) is performed by tactile stimulation of the cornea with a cotton-tipped applicator to elicit retraction of the ocular globe allowing extrusion of the third eyelid.
The parasympathetic component of CN VII mediates tearing, which is evaluated with Schirmer test strips.
Temporal and masseter muscle bulk and symmetry are assessed by observation (Fig. 10.20) and palpation; muscle tone is evaluated by opening the mouth (CN V–mandibular). With

Fig. 10.19. Tactile stimulation of the nasal septal mucosa tests the ophthalmic branch of the trigeminal (CN V) nerve and the contralateral forebrain. Even obtunded and stoic animals respond by retracting the head if the pathways underlying this response are intact. This test should be performed bilaterally and responses of each side should be compared. This test is particularly helpful to detect facial hypo-algesia in animals with forebrain disease. With lateralized forebrain disease, facial hypo-algesia can be detected on the side of the face contralateral to the affected cerebral hemisphere.
the animal’s mouth open, symmetry and movements of the tongue (CN XII) can be observed.
The gag reflex (CN IX, X, XI-internal branch) can be elicited by stimulating the pharynx with a finger or by applying external pressure on the hyoid bone. CN XI-external branch provides motor innervation to the trapezius and part of the sterno- and bachiocephalicus muscles. Focal lesions of this nerve are very rare and focal atrophy of these muscles may be difficult to appreciate clinically.
Hearing (receptors in the organ of Corti, CN VIII-cochlear branch, cochlear nuclei, pathways to auditory cortex) can be evaluated clinically by observing the animal orienting its heads and ears toward a loud or unexpected noise, such as a squeaky toy or whistle, or to the owner calling its name. Unilateral, or incomplete, deafness is difficult or impossible to detect clinically.
The conscious perception of smell (CN I, olfactory pathways to olfactory cortex) is difficult to assess objectively and therefore it is rarely evaluated during the neurological examination. Behavioural reactions to odours are controlled by connections to the limbic system. After the animal’s eyes have been covered, a small morsel of canned food is presented beneath the nose, observing for normal sniffing behaviour. Irritating substances, such as ammonia, should not be used, because they stimulate trigeminal nerve endings in the nasal passages and produce false results. Hyposmia or anosmia (decreased or absent sense of smell, respectively) are more commonly caused by non-neurological (e.g. rhinitis) than neurological disease. However, hypoanosmia can occur in animals with forebrain structural disease and seizures as the only obvious clinical abnormality.

Postural reactions
Postural reactions assess the same pathways involved in posture and gait (Fig. 10.21).

Fig. 10.21. Schematic illustration of the pathways tested during postural reaction assessment. Proprioceptive receptors in the joints detect abnormal paw position, the information is transmitted though the sensory component of the peripheral nerve, the spinothalamic pathway within the spinal cord, decussates in the brainstem and reaches the contralateral somatic sensory cerebral cortex. The information is relayed to the motor cortex with subsequent activation of descending motor pathways within the brainstem (which also decussate) and spinal cord (UMN), the motor component of the peripheral nerve, and skeletal effector muscles of the stimulated limb.
In addition, visual placing tests assess the visual pathways to the cerebral cortex, communication from the visual cortex to the motor cortex, and motor pathways to the LMN of the forelimbs. The main value of postural reaction testing is detection of subtle deficits or inconspicuous asymmetry that may not be obvious during the observation of gait. This is particularly valuable in animals with forebrain disease whose gait can be normal whereas postural reactions (particularly the paw replacement reaction) may be decreased. Postural reactions are also useful in discriminating between orthopaedic and neurological disorders. The role of postural reactions in achieving the neuroanatomic diagnosis is dependent on the rest of the neurologic examination. The animal should stand on a non-slip surface.
The paw replacement reaction is evaluated by supporting the animal’s body weight and turning each paw so that the dorsal surface is in contact with the floor (Fig. 10.22). The normal response is to immediately return the paw to a physiologic position.
The tactile and visual placing responses can be performed in small- to medium-sized dogs and in cats. The tactile placing test is performed first. The animal’s eyes are covered, the animal is picked up and moved toward the table so that the paw contacts the table’s edge. Normal animals immediately place the limb forward to rest the paw on the table surface. Individual limbs, both thoracic limbs, or the thoracic and pelvic limbs of each side can be tested (Fig. 10.23). Responses on the left and right side are compared. Some pets may not attempt to place the paw on the table as they are accustomed to being held. These animals should be held away from the examiner’s body as the resulting insecurity may persuade them to make an effort to place the limb on the table.
Visual placing is tested similarly, except the animal is allowed to see the table. Normal animals will place the paws on the table surface as the table is approached, before the paw touches the table. When unilateral visual deficits are suspected, each eye can be assessed individually by covering the contra-lateral eye.
The hopping reaction is tested by holding the animal so that the majority or all of its body weight is placed on the tested limb while the animal is moved laterally (Fig. 10.24). The other three limbs may be held off the ground, or the contralateral limb can be held off the


Fig. 10.24. The hopping reaction is tested in the left thoracic limb in this dog. The right thoracic and both pelvic limbs are lifted off the ground so that the majority of body weight is supported by the left thoracic limb. The dog is gently pushed to the left. The normal response is a smooth and rapid hopping movement to replace the examined limb under the body as the centre of gravity is moved laterally. This test should be performed on a non-slip surface.
ground while the rest of the body is supported. The normal response is a smooth and rapid hopping movement to replace the limb under the body as the centre of gravity is moved laterally. Each limb is tested individually, and responses on the left and right are compared.
Hemistanding and hemiwalking reactions are tested by lifting the thoracic and pelvic limbs on one side from the ground, so that all of the animal’s body weight is supported by the opposite limbs (Fig. 10.25). The animal is moved laterally away from the supported side. The normal response is similar as described for hopping. Responses on the left and right side are compared. This test can be particularly useful to detect deficits caused by lateralized forebrain pathology, which result in a decreased response in the contralateral limbs.
The wheel-barrowing reaction in the thoracic limbs is tested by supporting the animal under the abdomen so that the pelvic limbs do not touch the ground and moving the animal forward. Normal animals walk with symmetric, coordinated alternate movements of the thoracic limbs. Extending the head and neck eliminates visual compensation and allows detecting subtle proprioceptive deficits. The pelvic limbs can be tested similarly

Fig. 10.25. Hemistanding and hemiwalking reactions are tested in the right thoracic and pelvic limbs in this dog. The left thoracic and pelvic limbs are lifted off the ground so that the majority of body weight is supported by the right thoracic and pelvic limbs. The animal is gently pushed away from the side in which his limbs are supported. The normal response is a smooth and rapid hopping movement to replace each limb under the body as the centre of gravity is moved laterally. Responses on the left and right side are compared. This test should be performed on a non-slip surface and it can be particularly useful to detect deficits caused by lateralized forebrain pathology, which result in a decreased response in the contralateral limbs.
by supporting the animal under the thorax and moving the patient backwards.
The extensor postural thrust reaction is tested by supporting the animal by the thorax caudal to the scapulae. The animal is lifted and then lowered so that the pelvic limbs reach to the floor. The normal animal extends its limbs to contact the floor and steps backward with symmetric and coordinated walking movements.
Muscle mass and tone
All muscle groups should be systematically palpated, starting with the head, extending down the neck and the trunk, and continuing down each limb. Appendicular muscle mass and tone can be evaluated with the animal standing as well as in lateral or dorsal recumbence. The muscles are gently palpated and each limb is flexed and extended to evaluate muscle tone as well as joint range-of-motion. A decreased resistance to passive movement indicates reduced muscular tone (hypotonia) suggesting a LMN lesion. An increased resistance to passive movement indicates increased muscle tone (spasticity) suggesting an UMN lesion. Focal muscle atrophy developing in a week or two suggests a LMN lesion.
Spinal nerve reflexes
Examination of the spinal reflexes assesses the integrity of the sensory and motor components of the reflex arc and the influence of descending motor pathways on the reflex. A decreased or absent reflex generally indicates a partial or complete dysfunction, respectively, of either sensory or motor (LMN) components of the reflex arc. Transient hypo- or hypa-reflexia can occur caudal to the level of acute spinal cord injury in paralysed animals with ‘spinal shock’. This is followed by normoor a-reflexia over time. A lesion in the UMN pathways cranial to the spinal segment involved in the reflex arc results in normal or exaggerated reflexes. A wider excursion of the reflex movement can also occur with hypotonia of the antagonistic muscles (e.g. patellar pseudohyper-reflexia in animals with weakness of the muscles innervated by the sciatic nerve). Spinal reflexes are usually normal in animals with normal gait and postural reactions. Accurate evaluation of myotatic reflexes requires a relaxed patient in lateral or dorsal recumbence and proper technique. The lowest stimulus that can elicit a reflex should be used. The most reliable myotatic reflexes in the thoracic and pelvic limbs are the extensor carpi radialis and the patellar reflex, respectively.
The extensor carpi radialis reflex (C7-T1) is elicited by striking with a reflex hammer the extensor carpi radialis muscle (just distal to the elbow) while the limb is supported under the elbow, with the elbow and the carpus flexed by approximately 90° (Fig. 10.26). The normal reflex is a slight extension of the carpus.
The patellar reflex (L4-L6) is evoked by striking the patellar ligament with a reflex hammer while the limb is supported under the thigh, with the stifle partially flexed (Fig. 10.27). The normal reflex is a single, quick extension of the stifle.
The withdrawal (flexor) reflex (C6-T2 for the thoracic limb; L4-S1 for the pelvic limb) is tested in uppermost extended limbs of the laterally recumbent animal by gently pinching the toes (Fig. 10.28). The intensity of the stimulus can be gradually increased if necessary. The normal reflex is flexion of all the joints in the examined limb. Concurrent orthopaedic disorders restricting joint range of motion must be considered when evaluating this reflex. Presence of the withdrawal (flexor) reflex does not indicate conscious perception of the noxious stimulus applied to the toes.

The perineal reflex (S1-S3-Cd1-Cd5) is elicited by lightly stroking or pinching the left and right perineum with a haemostat. The normal reflex is contraction of the anal sphincter muscle and flexion of the tail.
The cutaneous trunci reflex is evaluated by pinching the skin of the thoracolumbar region between L5 and T2 vertebrae and observing a twitch of the skin resulting from the contraction of the cutaneous trunci muscles bilaterally. The afferent component is the spinal nerve of the stimulated dermatome; the sensory information ascends within the spinal cord to synapse bilaterally at the C8-T1 spinal cord segments with the lateral thoracic nerve LMNs which course through the brachial plexus and innervate the cutaneous trunci muscle. This reflex can be decreased or absent caudally to a thoracolumbar spinal cord lesion or it may be absent in its entire length due to a lesion in the efferent component of

Fig. 10.28. The withdrawal (flexor) reflex is tested in uppermost extended limbs of the laterally recumbent animal by gently pinching the toes. The normal reflex is flexion of all the joints in the examined limb. The stimulus of the lowest intensity that can elicit a response should be used. Concurrent orthopaedic disorders restricting articular range of motion must be considered when evaluating this reflex.
the reflex (generally unilaterally). This reflex cannot be elicited in some normal animals.
Evaluation of nociception
Nociception is defined as perception of noxious stimuli. Nociception over the digits, limbs and the tail is evaluated in animals with severe motor dysfunction (e.g. plegia) or with suspected sensory neuropathies. Facial sensation is assessed during cranial nerve examination and cutaneous sensation over the thoracolumbar region is tested while eliciting the cutaneous trunci reflex. Detecting and mapping out any areas of nociception loss helps to establish the neuroanatomic localization and has significant prognostic value in animals with spinal cord or peripheral nerve injuries.
Cutaneous (superficial) nociception is tested using a two-step pinch technique (Bailey and Kitchell, 1987) over specific dermatomes
(e.g. areas of skin innervated by a specific spinal nerve) or autonomous zones (e.g. areas of skin innervated by only one specific peripheral nerve). A small fold of skin is gently grasped and lifted with a haemostat. When the animal is quiet, the force of the pinch is gradually increased until a response is elicited. This response may be reflex (e.g. a skin twitch or limb flexion as described for the withdrawal reflex) or behavioural, such as turning the head towards the stimulus, vocalizing or attempting to bite. The behavioural response indicates perception of the noxious stimulus and therefore that the ascending nociceptive pathways in the peripheral nerve, spinal nerve, nerve root, spinal cord and brainstem to the forebrain are intact.
Deep nociception is tested when superficial nociception is absent. The examiner’s fingers or a haemostat are used to apply gradually increasing compression over the digits or the tail vertebrae in order to stimulate nociceptors in the periosteum until a response is elicited. As for superficial nociception, the response may be reflex or behavioural. Only the latter indicates perception of the noxious stimulus. The animal’s emotional state and level of consciousness may alter results of nociception testing.
Palpation of the head, spine and muscles
Palpation allows the detection of swelling, atrophy, deviation of normal contour (e.g. spinal malformations or fracture/luxation, calvarial tumours), persistent bregmatic fontanelles, depressed or elevated skull fractures as well as painful regions. Animals that are in extreme pain may react regardless of where they are palpated and therefore localization of the source of pain may be challenging. Animals with intracranial disease sometimes show sign of head or ‘referred’ cervical hyperaesthesia.
Seizure and AEM-associated neurological deficits
Dogs and cats examined shortly after a seizure may present with neurological deficits (e.g. obtundation, general proprioceptive ataxia, delayed postural reactions or blindness) associated with:
In addition, obtundation, ataxia (particularly in the pelvic limbs) and sometimes also decreased postural reactions may be observed in dogs and cats as side effects of certain AEMs. Neurologic examinations should be repeated over time on these animals.
Neuroanatomic Diagnosis
The history and neurologic examination findings help to establish whether the animal is affected by a neurologic disorder and if so identify location and distribution (e.g. focal, multifocal or diffuse, lateralized or symmetrical) of the lesion thereby reaching a neuroanatomic diagnosis (Plate 27). A history of seizures indicates a forebrain localization. The presence of other neurological signs and associated neuroanatomic localization (e.g. lateralized forebrain or multifocal intracranial) affects the differential aetiologic diagnosis list and diagnostic investigation plan. Neurological signs Animals with disorders of the neuromusassociated with lesions in different intracranial cular system have normal mental status and structures and spinal cord segments are sum-behaviour. Clinical signs vary depending on marized in Tables 10.1 and 10.2, respectively. the lesion location, e.g. cranial or spinal nerve,
Table 10.1. Neurological signs caused by a lesion in the forebrain, brainstem or cerebellum.
Forebrain (cerebrum Brainstem (midbrain, and diencephalon) pons, medulla oblongata) Cerebellum
Mental status Altered level of consciousness. and behaviour Behavioural changes. Hemineglect syndrome with lateralized lesions
Posture and gait With lateralized lesions: head turn, pleurothotonus and compulsive circling towards the side of the lesion. Normal gait to mild paresis
Cranial nerves Facial hypoalgesia, decreased or absent menace response and vision with normal pupil size and PLR (contralateral to lateralized cerebral lesions). With diencephalic lesions affecting optic chiasm or optic tracts: blindness with dilated unresponsive pupils
Postural Decreased or absent in all four reactions limbs or in thoracic and pelvic limbs contralateral to the lesion
Muscle tone and Normal to increased in all four spinal reflexes limbs or in thoracic and pelvic limbs contralateral to the lesion
Nociception Body hypo-algesia bilateral or contralateral to the lesion
Palpation May have head or referred cervical hyperaesthesia. Possible generalized hyperesthesia with certain thalamic lesions
Other findings Seizures, clinical signs of dysfunction of the hypothalamo-hypophyseal system
Altered level of consciousness (obtundation, stupor or coma)
Head tilt with vestibular lesions.
General proprioceptive or vestibular ataxia.
Tetraparesis or hemiparesis ipsilateral (or contralateral with certain midbrain lesions) to the lesion. Decerebrate rigidity (with midbrain lesions)
Abnormalities of CN III to XII (ipsilateral to lateralized lesions)
Decreased or absent in all four limbs or in thoracic and pelvic limbs ipsilateral to the lesion
Normal to increased in all four limbs or in thoracic and pelvic limbs ipsilateral to the lesion
Body hypo-algesia bilateral or ipsilateral to the lesion
May have cervical hyperaesthesia
Respiratory and cardiac abnormalities, or vomiting (with vestibular disease) Normal
Wide-based stance, cerebellar or cerebello-vestibular ataxia, intention tremors.
Possible head tilt.
Decerebellate rigidity (with rostral cerebellar lesion)
Normal or possibly vestibular signs (head tilt, abnormal nystagmus), anisocoria and ipsilateral decreased or absent menace response with normal vision and facial nerve motor function
Delayed initiation and exaggerated response or normal
Normal to increased muscle tone
Normal
Normal
Possibly increased frequency of urination
Table 10.2. Neurological signs caused by lesion in various spinal cord segments.
C1-C5 C6-T2 T3-L3 L4-L6 L7-S3-Cd5
Level of consciousness and behaviour
Posture and gait
Cranial nerves Postural reactions
Muscle tone and spinal reflexes
Nociception
Palpation Other findings Normal
Low head carriage, torticollis, scoliosis, general proprioceptive ataxia, paresis/plegia of all four limbs or in thoracic and pelvic limbs ipsilateral to the lesion
Possible ipsilateral Horner’s syndrome
Decreased to absent in all four limbs or in thoracic and pelvic limbs ipsilateral to the lesion
Normal to increased in all four limbs or in thoracic and pelvic limbs ipsilateral to the lesion
Decreased to absent in tetra/hemi-plegic animals
Cervical hyperaesthesia
Respiratory difficulty in tetraplegic animals Normal
Low head carriage, torticollis, general proprioceptive ataxia, paresis/plegia of all four limbs or in thoracic and pelvic limbs or in thoracic limb ipsilateral to the lesion. Possible lameness in one or both thoracic limbs
Possible ipsilateral Horner’s syndrome
Decreased to absent in all four limbs or in thoracic and pelvic limbs or one thoracic limb ipsilateral to the lesion
Decreased to absent in one or both the thoracic limbs, normal to increased in the pelvic limbs. Decreased or absent cutaneous trunci reflex (ipsilateral to the lesion)
Decreased to absent in tetra/hemi/mono-plegic animals
Cervico-thoracic hyperaesthesia
Respiratory difficulty in tetraplegic animals Normal
Thoracolumbar kyphosis, scoliosis, general proprioceptive ataxia, paresis/plegia of one or both pelvic limbs; Shiff-Sherrington posture (with acute severe lesions)
Normal
Normal in the thoracic limbs, decreased to absent in one or both pelvic limbs
Normal in the thoracic limbs, normal to increased in one or both pelvic limbs. With ‘spinal shock’ may be transiently decreased in one or both pelvic limbs. Cutaneous trunci reflex decreased or absent caudally to the lesion
Decreased to absent in one or both pelvic limbs and tail
Thoracolumbar hyperaesthesia
Tail paresis/plegia, UMN bladder
Normal
Lumbar kyphosis, general proprioceptive ataxia, paresis/plegia of one or both pelvic limbs
Normal
Normal in the thoracic limbs, decreased to absent in one or both pelvic limbs
Normal in the thoracic limbs, decreased to absent patellar reflex in one or both pelvic limbs. Normal withdrawal reflex in the pelvic limbs. Quadriceps muscle weakness and possible atrophy
Decreased to absent in the L4-L5-L6 dermatomes
Lumbar hyperaesthesia
Tail paresis/plegia, UMN bladder Normal
Lumbosacral kyphosis, general proprioceptive ataxia, paresis or lameness of one or both pelvic limbs
Normal
Normal in the thoracic limbs, decreased to absent in one or both pelvic limbs
Normal in the thoracic limbs, normal patellar reflex or pseudo-hyperreflexia in both pelvic limbs; decreased to absent withdrawal reflex in one or both pelvic limbs and perineal reflex. Atrophy of the muscles supplied by the sciatic nerve
Decreased to absent in the L7-S1-3 dermatomes
Lumbosacral hyperaesthesia
Flaccid tail paresis/plegia, LMN bladder, dilated anal sphincter, faecal incontinence
Investigation of the Seizure Patient
297
sensory or motor component, neuromuscular junction, or muscle.
Peripheral nerve dysfunction generally is characterized by flaccid paresis or paralysis, decreased to absent postural reactions and spinal reflexes, and muscle hypotonia and atrophy. CN involvement results in various clinical signs depending on the affected CN (see cranial nerve examination). Nociception may be decreased or absent in the dermatome of the affected nerve. Hyperalgesia or paraesthesia can occur. Some neuropathies are exclusively or primarily characterized by motor dysfunction, others by sensory dysfunction and some by a combination of both motor and sensory dysfunction. With mononeuropathies, deficits are restricted to regions innervated by the affected nerve. Polyneuropathies may affect multiple spinal or cranial nerves or both.
Muscle disorders are often characterized by weakness, fatigability and stiff, stilted gait. Apostural tremor may be observed. Masticatory muscles, muscles of facial expression or pharyngeal and laryngeal muscles may also be involved. Regurgitation can occur with oesophageal skeletal muscle involvement. Muscle tone may be normal, increased or decreased. Spinal reflexes are usually normal, but may be weak or fatigable, or difficult to evoke in animals with severe muscle atrophy and fibrosis. Palpation may reveal muscle atrophy or less commonly hypertrophy, and sometimes hyperaesthesia. Sensations (including proprioception and nociception) are normal.
Animals with neuromuscular junction disorders may have a normal neurologic examination following rest, however various degrees and duration of exercise often results
Seizures
in fatigability and stiff, stilted gait which improves or resolves following rest. Postural reactions are normal, although profound weakness may affect performance. Spinal reflexes are usually normal but may be fatigable or weak. Muscles of facial expression, pharyngeal, laryngeal, oesophageal skeletal muscles may also be involved resulting in facial paresis, dysphonia, dysphagia and regurgitation.
Differential Aetiologic Diagnoses
The main variables to consider when developing the differential aetiologic diagnosis list in animals presenting with seizures are the following (Fig. 10.29):
The differential aetiologic diagnosis list, ordered from most likely to least likely diseases, represents an essential step in choosing and interpreting any diagnostic tests. Differential aetiologic diagnoses in animals presenting with seizures and associated veterinary seizure/epilepsy classification are listed in Table 3.3, Boxes 3.1 and 3.2 (see Chapter 3). Information on each aetiologic
Age onset between 6 months and 6 years Age onset <6 months or >6 years
Interictally normal Interictally abnormal Interictally normal Interictally abnormal
Idiopathic epilepsy Structural or reactive Structural, reactive or cryptogenic seizures seizures
Fig. 10.29. Flow chart of differential diagnosis in the seizure patient.
Table 10.3. Onset and course of neurological signs with different disease categories.
Disease category Disease onset Disease course Distribution
Vascular Peracute or acute
Inflammatory/ Acute, subacute infectious or chronic
Traumatic Peracute or acute
Toxic Acute; history of toxin exposure. Lead may cause chronic signs
Anomalous and Subacute or chronic, developmental generally early in life. Occasionally acute if loss of compensatory mechanisms in the adjacent normal tissue occurs
Metabolic Acute, or subacute, to chronic
Nutritional Acute, or subacute, to chronic
Neoplastic Subacute or chronic. Occasionally acute with associated haemorrhage, ischaemia or loss of compensatory mechanisms in the adjacent normal tissue
Degenerative Chronic
Non-progressive, regressive. Deterioration of one to few days duration can occur with progressive oedema or haemorrhage
Progressive deterioration, occasionally relapsing remitting course
Non-progressive, may improve with time. Deterioration of one to a few days duration can occur with progressive oedema or haemorrhage
Variable. Presence of concurrent signs due to involvement of other systems
Non-progressive or slowly progressive deterioration.
Littermates or other family members may have the same or similar signs
Progressive deterioration or relapsing remitting Progressive deterioration
Progressive deterioration
Progressive deterioration. With genetic neurodegenerative disorders, littermates or other family members may have the same or similar signs
Focal, often lateralized; may be multifocal with multiple haemorrhages
Focal, often lateralized
(e.g. abscess, granuloma, focal GME), diffuse or multifocal, lateralized or symmetric
Often focal and lateralized, sometimes multifocal
Diffuse and bilaterally symmetric
Focal, diffuse or multifocal, symmetric or lateralized
Diffuse and bilaterally symmetric Diffuse or multifocal and bilaterally symmetric Focal, often lateralized
Often diffuse and symmetric
Peracute, <6 h; acute, 7–24 h; subacute, onset over a few days; chronic, onset over several days, weeks or months. Anatomic distribution of a disorder (e.g. focal, multifocal/diffuse, symmetric/lateralized) does not always correspond to distribution of clinical signs. Disorders that typically result in bilateral symmetric diffuse forebrain or intracranial disease (e.g. metabolic diseases) may present in compulsive circling suggesting lateralized disease. Similarly, focal lateralized lesions (e.g. cerebral abscess, granuloma, neoplasia or haemorrhage) may have extensive perilesional oedema and associated mass effect resulting in diffuse lateralized rather than focal neuroanatomic diagnosis.
diagnosis including clinical presentation, diagnostic investigations, treatment and prognosis are presented in Chapters 4, 5 and 6.
The main differential diagnosis in dogs presenting with epilepsy (e.g. recurrent seizures) as the sole abnormality, seizure onset between 6 months and 6 years of age, and no abnormalities detected on repeated physical and neurological examinations is idiopathic epilepsy (see Chapter 6). Breed predisposition (see Table 6.1) makes idiopathic epilepsy even more likely. In a retrospective study on a referral population of 240 seizuring dogs, seizure onset between 1 and 5 years was associated with a 3.25 times greater likelihood for idiopathic epilepsy than symptomatic (structural) epilepsy and reactive seizures (Pákozdy et al., 2008). Similarly, in a study on a non-referral canine population, symptomatic (structural) epilepsy was statistically more probable in dogs <1 year or >7 years of age (see Podell et al., 1995; Chapter 3). The absence of interictal abnormalities, however, does not completely rule out structural epilepsy as focal lesions in particular areas of the forebrain (‘clinically silent regions’), such as olfactory bulb and frontal lobes, can result in seizure activity without any other neurological signs (Fig. 10.30a–d) (see Chapter 3; Foster et al., 1988; Smith et al., 1989; Smith et al., 2008). Indeed, a considerable proportion (53/90, 59%) of dogs ³7 years of age with an unremarkable neurologic examination had an identifiable underlying structural CNS disease (Schwartz et al., 2013).
The presence of interictal forebrain signs unrelated to the post-ictal period, seizure-induced hypoxic or excitotoxic cerebral injury or AEM side-effects suggests an underlying structural, metabolic, nutritional or toxic brain disease.
Diagnostic Investigations
Diagnostic investigations must be selected and interpreted based on the neuroanatomic diagnosis and differential aetiologic diagnosis list. Diagnostic investigations and possible indications in animals presenting with seizures are summarized in Table 10.4. Diagnostic investigations for specific aetiologies of reactive seizures and structural (symptomatic) epilepsy as well as to support the diagnosis of idiopathic epilepsy are detailed in Chapter 4, 5 and 6, respectively. Idiopathic epilepsy refers to recurrent seizures with no underlying cause other than a strongly suspected or confirmed genetic or familial basis. In the absence of a genetic test, the diagnosis is currently based on exclusion of metabolic, toxic and structural cerebral disorders by means of careful history-taking, physical and neurological examinations and diagnostic investigations.
Haematology, serum biochemistry and urinalysis
Complete blood count, comprehensive serum biochemistry (including fasting glycaemia, electrolytes and fasting bile acids) and urinalysis should be performed as minimum database in all dogs and cats with a history of one or more seizures. Results of minimum database tests may be normal. However, they may reveal the following:
Hepatic enzymes may be increased shortly after seizure activity because of the effects of hypoxia and hypotension.
The minimum database tests also provide information on general health prior to general anaesthesia, which may be necessary for further investigations (e.g. MRI and CSF) and before initiation of anti-epileptic treatment. The reader is referred to a clinical pathology

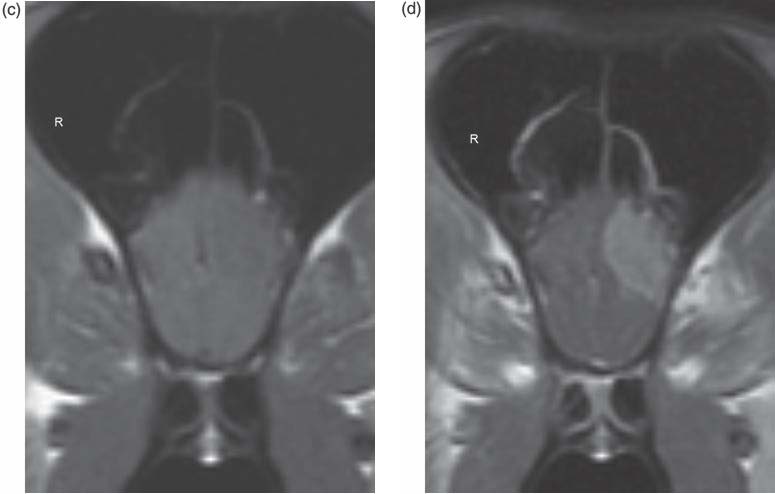
Fig. 10.30. MRI of a 9-year-old female spayed German shepherd dog with recent onset of cluster seizures and no abnormalities on neurological examination. Paramedian T2W (a), transverse T1W (c) and T1WC (d) images at the level of the olfactory lobe and dorsal T1WC (b) show a well marginated ovoid extra-axial mass, located within the left olfactory and rostral frontal lobes. The mass is hyperintense to grey matter on T2W, isointense on T1W images and demonstrates diffuse contrast enhancement. A transitional meningioma was diagnosed histologically following surgical excision.
Table 10.4. Indication for various diagnostic investigations in animals presenting with seizures.
Diagnostic investigation Indication
| Haematology, serum biochemistry and | All dogs and cats with a history of one or more seizures |
| urinalysis (minimum database) | |
| Fasting plasma ammonia concentration | Suspected hepatic encephalopathy |
| Pre- and post-prandial bile | Suspected hepatic encephalopathy |
| acid concentration | |
| Endocrine testing | Suspected endocrinopathies (e.g. insulin levels obtained |
| when glucose concentration is <60 mg/dl in dogs with | |
| suspected insulinoma) | |
| Tests of haemostasis | Suspected haemorrhagic diatheses |
| Toxicological testing | Suspected neurotoxicity based on history of possible toxin |
| exposure, clinical examination and minimum database findings | |
| Radiography of the thorax | Suspected neoplastic or certain infectious disorders (i.e. fungal |
| and bacterial infections, pneumocystis, Toxoplasma gondii) | |
| Abdominal ultrasonography | Suspected portosystemic shunt or hepatic disease, renal |
| disease, adrenal disease, insulinoma or other systemic | |
| metabolic abnormality suggested by the minimum | |
| database; suspected primary or metastatic neoplasia | |
| Magnetic resonance imaging | To investigate possible structural brain disorders |
| (e.g. vascular, infectious/ inflammatory, traumatic, | |
| anomalous, neoplastic, degenerative). Helpful to support | |
| the diagnosis of thiamine deficiency | |
| To support the diagnosis of idiopathic epilepsy | |
| CSF analysis (cell count, total and | To investigate possible infectious/inflammatory CNS disorders |
| differential; protein concentration) | and certain neoplastic disorders (e.g. lymphoma) |
| To support the diagnosis of idiopathic epilepsy | |
| Infectious disease testing | Suspected meningoencephalitis based on history, clinical, |
| minimum database, MRI and CSF analysis findings |
Quantification of amino acids and organic Suspected inborn errors of metabolism (Sargan, 2007) acids and determination of glycosaminoglycans, oligosaccharides, purines and pyrimidines in the serum, CSF or urine
Genetic testing Suspected genetic disorder with known mutation as seizure cause (see Table 10.8) Electroencephalography Confirmation of seizure activity
or internal medicine book for in-depth discussion of various laboratory abnormalities and associated differential diagnoses.
Hepatic function testing
Fasting plasma ammonia and fasting (e.g. 12-h) and post-prandial (e.g. 2-h) total serum bile acid concentrations should be assessed in dogs and cats with suspected hepatic encephalopathy (see Chapter 4) and to obtain a thorough baseline assessment of hepatic function in any animal that may need to be treated with hepatically metabolised and/or potentially hepatotoxic AEMs such as phenobarbital.
Toxicological testing
Toxicological testing should be performed any time neurotoxicity is the suspected cause of the seizures. The suspicion of toxin exposure is often based on the history and/or the acute onset of seizures in combination with other neurological signs (including excitation and hyperactivity or obtundation, stupor or coma, muscle tremors and fasciculations, and ataxia) preceded or accompanied by vomiting, diarrhoea, salivation, bronchoconstriction, bradycardia or tachycardia, and hyperthermia. Chronic exposure to lead can result in recurrent seizures as the sole or predominant clinical abnormality. When the
Table 10.5. Minimum database abnormalities associated with selected underlying seizure aetiologies and specific further investigations required to confirm the diagnosis.
Minimum database Suspected underlying Required further abnormalities seizure aetiologies investigations
Hypoalbuminaemia, hypoglycaemia, decreased blood urea nitrogen, hypocholesterolaemia, +/− increased hepatic enzyme and total bilirubin; microcytosis with or without hypochromic anaemia; ammonium biurate crystals in the urine sediment
Hypoglycaemia
Atypical lymphocytosis or lymphocytic leukaemia
Inflammatory leucogram
Inflammatory leucogram, hyperglobulinaemia
Nucleated erythrocytes, basophilic stippling, +/− mild normocytic, normochromic, non-regenerative anaemia
Azotaemia, hypocalcaemia, hyperkalaemia, hyperphosphataemia, hypernatraemia, marked hyperglycaemia (in cats), anion gap >40–50 mEq/l, metabolic acidosis; hyposthenuria, calcium oxalate monohydrate crystalluria
Hepatic encephalopathy (portosystemic shunt, severe hepatic disease)
Insulinoma
CNS lymphoma
Bacterial or fungal meningoencephalitis
Feline infectious peritonitis
Lead toxicity
Ethylene glycol toxicity
Fasting serum ammonia concentration, fasting and postprandial bile acid concentrations, abdominal ultrasound, +/− portovenography, scintigraphy (transcolonic or trans-splenic), computed tomographic angiography or magnetic resonance angiography, hepatic biopsy
Insulin levels obtained when glucose concentration is <60 mg/dl, abdominal US, abdominal CT, cytology, histology
Thoracic radiography, abdominal US, lymph node or lesion cytology or histology, brain MRI, CSF analysis
Antigen titres, PCR, MRI, CSF analysis, microbial culture and susceptibility, thoracic radiography, abdominal US
Serum protein electrophoresis, thoracic radiography, abdominal US, cytology, brain MRI, CSF analysis, PCR, detection of FCoV antigen within macrophages in body fluids, cytology or biopsy specimens
Lead blood levels ≥40 µg/dl
Ethylene glycol colorimetric spot tests (urine or serum); serum, plasma or urine laboratory analysis for ethylene glycol and glycolic acid
source of intoxication is known or highly suspected, toxicological testing can be focused on specific toxins (e.g. lead, metaldehyde, strychnine, penitrem A and roquefortine). Feed or suspect bait material, gastric content from vomitus or lavage fluids, water, blood or urine (for urinary-excreted toxins) can be submitted to the laboratory for analysis of suspected neurotoxins or toxicological screen (when available). Samples may need to be kept frozen for identification of certain toxins
(e.g. metaldehyde, zinc phosphide). Exposure to organophosphates in dogs can be confirmed by identification of a decreased blood cholinesterase activity by 50% or more of normal. Ethylene glycol intoxication can be confirmed by colorimetric spot tests or laboratory tests on urine and serum. Spot tests can give false negatives in cats as the ethylene glycol toxic dose in cats can be below the detectable level of the ethylene glycol test kit (Smith and Lang, 2000; Van Hee et al., 2004). Certain neurotoxins such as bromethalin can be identified only in body tissues and therefore the diagnosis can be reached only post-mortem. Toxicological testing available for specific toxins are detailed in Chapter 4.
Survey radiography of the thorax and ultrasography of the abdomen
Survey radiography of the thorax and radiography or ultrasonography of the abdomen should be performed in animals with suspected neoplastic (Fig. 10.31) or systemic infectious disease (e.g. fungal). Abdominal ultrasound is also indicated to investigate certain metabolic disorders causing seizures such as insulinoma-associated hypoglycaemia (see Fig. 4.1, Chapter 4) and hepatic encephalopathy due to a portosystemic shunt (see Fig. 4.3, Chapter 4).
Magnetic resonance imaging
Magnetic resonance imaging (MRI) is the diagnostic imaging modality of choice for evaluation of the brain in animals with seizures. MRI allows imaging in any anatomical plane and provides excellent soft-tissue contrast resolution. A minimum database and if appropriate other investigations to rule out reactive seizures are performed prior to MRI. MRI is indicated any time structural (symptomatic) epilepsy is suspected (e.g. interictal neurological abnormalities or age at seizure onset <6 months or >6 years) and to support the diagnosis of idiopathic epilepsy. MRI is also indicated following exclusion of reactive seizure aetiologies, in any cat with recurring seizures as structural (symptomatic) epilepsy is common in cats and, unlike dogs, a breed-specific predisposition for idiopathic epilepsy has not been identified yet (Quesnel et al., 1997; Cizinauskas et al., 2003; Schriefl et al., 2008). In addition, MRI can help to support a timely diagnosis of thiamine deficiency due to characteristic lesion distribution and signal changes (see Chapter 4).

The presence of interictal neurological abnormalities and the animal’s age have been evaluated in attempt to predict the probability of identifying structural cerebral disorders with MRI in epileptic dogs. One study reported brain MRI abnormalities in 22% (14/63) and 90% (47/52) of epileptic dogs with normal and abnormal neurological examination, respectively (Bush et al., 2002). Results of CSF analysis (normal versus abnormal) were significantly associated with results of MRI (normal versus abnormal) in both dogs with normal and abnormal neurological examination. In this study, altered mental status and signs of cervical hyperesthesia were not considered abnormal neurological findings. Age of the dog at seizure onset (<6 versus ³6 years) was not significantly associated with brain MRI results (normal versus abnormal) for any groups when dogs were grouped based on neurological examination and CSF analysis results (normal versus abnormal) (Bush et al., 2002). Another study reported clinically significant MRI abnormalities including olfactory or frontal lobe neoplasia in 2.2% (1/46) and 26.7% (8/30) of interictally normal epileptic dogs younger and older than 6 years of age, respectively (Smith et al., 2008). In a study including dogs whose first seizure occurred below the age of 1 year, 26% (6/23) of dogs with a normal neurological examination had an underlying structural brain disease identified with MRI and CSF analysis (Arrol et al., 2012). A recent study including dogs whose first seizure occurred ³7 years of age identified an underlying CNS structural disease in 59% (53/90) of dogs with an unremarkable interictal neurologic examination (Schwartz et al., 2013).
The physical basis and techniques for performing MRI are beyond the scope of this book and the reader is referred to other textbooks and reviews (Hecht and Adams, 2010; Robertson, 2011). MRI sequences are designed to acquire information by exploiting differences in behaviour of hydrogen protons in various tissues in changing magnetic fields (Hecht and Adams, 2010). Commonly used MRI pulse sequences for brain imaging include spin echo T1-weighted pre-contrast (T1W) and post-contrast (T1WC), T2-weighted (T2W), T2weighted fluid attenuated inversion recovery (FLAIR) and T2*-weighted gradient echo (T2*W GRE) images (Fig. 10.32a–e). Other sequences including diffusion-weighted imaging (DWI) (Figs 5.1e, 5.2e), perfusion-weighted imaging (PWI), fat suppression techniques (short tau inversion recovery (STIR) and chemical fat suppression (fat saturation (Fat Sat)) are used to investigate specific disorders (Hecht, 2010). The indication and main advantages of MRI pulse sequences for brain imaging are summarized in Table 10.6. The signal intensities of fat, CSF and tissue oedema in different MRI pulse sequences are presented in Table 10.7.
MRI field strength, image acquisition protocols, interpretation technique and expertise affect the ability to recognize cerebral pathology in epileptic patients.
MR images are assessed mainly for changes in symmetry of anatomical structures and alterations in signal intensity of brain parenchyma. Suspect lesions should be cross-referenced with different imaging planes and sequences. Comparison of signal intensity of lesions on different pulse sequences allows evaluating the properties of the tissue. Knowledge of cerebral anatomy including normal anatomic variations and awareness of imaging artefacts are important when interpreting MRI (Cooper et al., 2010). The MRI features of various aetiologies of structural (symptomatic) epilepsy have been described in Chapter 5 and have been reviewed elsewhere (Hecht and Adams, 2010a, b; MacKillop, 2011; Rodenas et al., 2011; Wisner et al., 2011; Young et al., 2011). Sensitivity and specificity of MRI in the diagnosis of neoplastic, inflammatory and cerebrovascular brain diseases have been reported (Cervera et al., 2011; Wolff et al., 2012).
General anaesthesia is required to perform MRI and represents the main risk associated with this procedure. The animal should have an easily accessible, patent intravenous



catheter, be intubated and if necessary ventilated, and adequately monitored (e.g. heart and respiratory rate, electrocardiogram, saturation of peripheral oxygen, end tidal carbon dioxide and body temperature). Adequate blood pressure and oxygenation should be maintained during MRI.
Seizure-associated MRI changes
Severe seizure activity has been reported to cause reversible MR signal changes in certain areas of the brain, such as the temporal and frontal cortex, hippocampus and amygdala in people and dogs (Chan et al., 1996; Aykut-Bingol et al., 1997; Mellema et al., 1999; Kim et al., 2001; Briellmann et al., 2005; Huang et al., 2009). These signal changes are characterized by varying degrees of hyperintensity on T2-weighted, FLAIR and diffusion-weighted imaging, reduced apparent diffusion coefficient (ADC), hypointensity on T1-weighted images, and occasionally heterogeneous contrast enhancement following gadolinium administration (Mellema et al., 1999; Kim et al., 2001; Huang et al., 2009). The location of these MRI changes corresponds to areas of maximal EEG abnormality in people (Cole, 2004). In dogs, these changes have been identified unilaterally or bilaterally, predominantly in the piriform and temporal lobes (Fig. 10.33), but also in the olfactory bulb and frontal lobe on MRI performed within 14 days of the last seizure. Following anti-epileptic treatment only, these signal changes partly or completely resolved on repeated MRI 10 to 16 weeks after the initial one, indicating that they most probably represent cytotoxic and vasogenic oedema induced by seizures. Histologic examination of the affected temporal cortex, hippocampus and piriform lobe reveals oedema, neovascularization, reactive astrocytosis and acute neuronal necrosis. Repeated MRI after a period of seizure control, along with clinical and CSF analysis findings, may help to differentiate
Table 10.6. MRI pulse sequences for brain imaging.
Pulse sequence Indication Advantage
| T1-weighted (pre- | Every brain MRI protocol |
| and post-contrast) | |
| T2-weighted | Every brain MRI protocol |
| T2-weighted FLAIR | Every brain MRI protocol, |
| suspected CNS inflammatory | |
| disease, hydrocephalus, cystic | |
| lesions | |
| T2*-weighted (GRE) | Every brain MRI protocol, |
| suspected haemorrhage | |
| Diffusion-weighted | Suspected hyperacute cytotoxic |
| imaging | oedema |
| Fat suppression | Further characterization of |
| techniques | lesion within or adjacent to fat |
| (STIR, Fat Sat) | tissue |
Good anatomical detail. Comparison of pre- and post-contrast studies allows evaluation of lesion vascularization and blood-brain barrier disruption
Very sensitive in detecting pathology
Allows suppression of CSF signal and helps to characterize T2 hyperintense lesions. May identify small lesions bordering a fluid-filled ventricle or subarachnoid space, meningeal disease, small infarcts or inflammatory lesions more clearly than T2-weighted images. Post-contrast FLAIR images are very sensitive in the detection of contrast-enhancing lesions.
Can help to visualize seizure-induced cytotoxic and vasogenic oedema
Most sensitive sequence to detect intraparenchymal haemorrhage
(e.g. haemorrhagic infarcts, coagulopathies, haemorrhagic metastasis or angiostrongylosisassociated haemorrhage)
Identification of peracute cerebral infarcts.
Can help to visualize seizure-induced cytotoxic oedema
Allows suppression of fat signal and visualization of lesions within or adjacent to fat tissue
MRI, magnetic resonance imaging; FLAIR, fluid attenuated inversion recovery; GRE, gradient echo; STIR, short tau inversion recovery; CNS, central nervous system; CSF, cerebrospinal fluid
Table 10.7. Signal intensity of fat, CSF and tissue oedema in different MRI pulse sequences (intensity is described relative to normal cerebral grey matter).
Tissue Pulse sequence Fat CSF oedema
| T1-weighted (pre- and | Hyperintense | Hypointense | Hypointense |
| post-contrast) | |||
| T2-weighted | Hyperintense | Hyperintense | Hyperintense |
| T2-weighted FLAIR | Hyperintense | Hypointense | Hyperintense |
| T2*-weighted | Hyperintense | Hyperintense | Hyperintense |
| Fat suppression techniques | Hypointense | Hyperintense | Hyperintense |
| (STIR, Fat Sat) |
seizure-induced changes from inflammatory changes may evolve into permanent abnoror neoplastic epileptogenic structural lesions malities such as focal neuron cell loss and (Mellema et al., 1999; Pákozdy et al., 2013). gliosis potentially leading to an epileptogenic However, seizure-induced acute parenchymal lesion visible on MRI (Briellmann et al., 2005).


Diffusion tensor imaging
Diffusion tensor imaging (DTI) is a novel MR technique, which has evolved from diffusion-weighted imaging (Plate 28). DTI measures diffusion properties of water protons in tissue and can detect subtle white-matter changes in different pathological states including epilepsy (Basser and Pierpaoli, 1996; Chahboune et al., 2009). DTI is a sensitive method for detecting abnormalities in patients with focal and generalized epilepsies, even in structures without apparent changes on conventional MRI (Kimiwada et al., 2006). In addition, DTI can help to study changes in the underlying connectivity of the epileptic brain (Concha et al., 2009, 2010). DTI data can be displayed in a three-dimensional format referred to as fibre tractography, which allows illustration and visualization of specific tracts such as connections with the optic radiation or language cortex in people. This imaging modality can help predict language or visual deficits that may result from temporal lobe resection (Powell et al., 2005, 2008).
Magnetic resonance volumetry
MRI volumetry and morphometry are used to compare the size and shape of brain structures. Volumetric measurements can be done using a variety of methods. The most frequently used is voxel-based morphometry (VBM), which is done by spatially normalizing all images, segmenting grey matter from images, and then performing voxelwise parametric statistical tests to produce a parametric map of structural regions (Ashburner and Friston, 2000). MRI volumetry can detect decreases in volumes of structures functionally connected to the hippocampus, such as the amygdala, entorhinal cortex, fornix, mamillary body and thalamus and help to study changes in these structures during epileptogenesis. The volume loss appears related to the duration of epilepsy and may be associated with a pre-existent injury or result from recurrent seizures (Seidenberg et al., 2005; Szabó et al., 2006).
Magnetic resonance volumetry of the hippocampus has been investigated in dogs using different magnetic field strengths and sequences (e.g. proton-density-weighted, T2-weighted, 3-D MPRAGE and T1-weighted 3-D ultrafast gradient echo) (Vullo et al., 1996; Jung et al., 2010; Kuwabara et al., 2010; Milne et al., 2013). The most recently described volumetric technique uses a 1.5-Tesla MRI scanner to acquire a T1-weighted 3-D ultrafast gradient echo sequence aligned in an oblique dorsal plane perpendicular to the long axis of the hippocampus (Fig. 10.34a, b). The plane of acquisition needs to be determined on a

Fig. 10.34. Oblique dorsal T1-weighted 3-D ultrafast gradient echo sequence (a) acquired on a plane perpendicular to the long axis of the hippocampus (which has been initially determined on a sagittal T2W FSE image). Parasagittal T1-weighted 3-D ultrafast gradient echo image (b) reformatted from the dorsal plane acquisition. Note the hippocampal formation between the arrows and the plane of acquisition of the dorsal oblique image (dashed line) (photo courtesy of Marjorie Milne and Sam Long, University of Melbourne Veterinary Hospital).
patient-by-patient basis, due to individual for the right and left, respectively, canine hip-variation in orientation of the canine hip-pocampal volumes adjusted for patient size pocampal formation. The hippocampal volume on the basis of intracranial volume has been is measured with imaging software (Osirix) established (Milne et al., 2013). These refer-after manual delineation of the hippocampus. ence values could help detecting hippocam-A lower reference limit of 0.56 cm3 and 0.55 cm3 pal atrophy in epileptic dogs.

Fig. 10.35. Multi-slice voxel magnetic resonance spectroscopy performed on a normal canine brain. Each voxel reveals a small graphical representation of the metabolites present within its region of the brain. This type of spectroscopy allows analysis of small regions of the brain. Computer software analysis will define the peaks of the metabolites objectively.
Functional neuroimaging
In addition to structural MRI, investigation of epilepsy in people often involves functional neuroimaging by magnetic resonance spectroscopy (MRS), positron emission tomography (PET), single-photon emission computed tomography (SPECT) and functional MRI (fMRI) (Mishra et al., 2011). These functional imaging modalities have been used in animal models of epilepsy (Neppl et al., 2001; Powell et al., 2001; Hiremath and Najm, 2007; Goffin et al., 2008; Mirrione and Tsirka, 2011) and their clinical application in veterinary medicine may revolutionize our ability to assess and treat epileptic animals.
Magnetic resonance spectroscopy
Magnetic resonance spectroscopy (MRS) and the related technique of magnetic resonance spectroscopic imaging (MRSI) are used in both clinical and preclinical research as well as in clinical practice for the non-invasive evaluation of brain metabolism (Zhu and Barker, 2011). MRS can be performed in a single integrated examination with conventional high-field MRI. MRS detects magnetic resonance signals produced by hydrogen (1H or proton MRS) or phosphorus (31P MRS) atomic nuclei to estimate the relative concentrations of target brain metabolites in the region of interest. MRS of carbon (13C), sodium (23Na) and fluoride (19F) atomic nuclei can be performed with MRI of 3 Tesla or higher magnetic field strength. 1H or proton MRS is the most commonly used type of MRS (Fig. 10.35). Proton MR spectra can be acquired using either a long echo time (TE >100 ms) or a short echo time (TE 20–35 ms). Long-echo acquisitions produce spectra provide information about N-acetyl aspartate (NAA), choline-containing metabolites (Cho), creatine/phosphocreatine (Cr) and lactate (Lac). Short-echo spectra provide information about NAA, Cr, Cho and Lac as well as glutamate and glutamine (Glx), myoinositol (mI), aspartate and alanine, and several other metabolites. NAA is a neuronal and axonal marker that decreases with neuronal loss or dysfunction; Cr is a marker for intact brain energy metabolism; Cho is a marker for membrane synthesis or repair, inflammation or demyelination; Lac is a metabolite of anaerobic glycolysis; mI is a glial marker; and Glx is an epileptogenic excitatory neurotransmitter (Ross and Bluml, 2001). Reduction of the ratio of NAA to choline, creatine and phosphocreatine is a marker of neuronal loss and dysfunction. 31P MRS is the second most common type of MRS and can detect adenosine triphosphate, phosphodiesters, phosphomonoesters, phosphocreatine and inorganic phosphate, and estimate intracerebral pH.
MRS allows detecting metabolic abnormalities in seizure foci and areas of seizure spread (Najm et al., 1998; Simister et al., 2008, 2009). MRS metabolite abnormalities may be identified even in the absence of detectable structural abnormalities on conventional MRI (Connelly et al., 1998). The ratio (NAAt/Cr) between N-acetyl aspartate plus N-acetyl aspartyl-glutamate (NAAt) and creatine plus phosphocreatine (Cr) was reduced in ipsilateral and contralateral temporal lobes in people with temporal lobe epilepsy (TLE) attributable to unilateral hippocampal sclerosis (Simister et al., 2009). Normalization of NAAt/Cr in the contralateral temporal lobe was observed following successful anterior temporal lobe resection (Simister et al., 2009). A reduction of NAA content and unilateral presence of Lac in the mesial temporal lobe structures has been reported in people with mesial temporal lobe epilepsy (mTLE). This metabolic abnormality contributed to identify the lateralization of the epileptogenic zone. Significant bilateral metabolic alterations in the mesial temporal lobe structures were associated with worse post-operative seizure control (Chernov et al., 2009). Proton MRS abnormalities in people with idiopathic generalized epilepsy include elevated glutamine concentrations (suggesting increased neuronal excitability) and reduction in NAAt concentrations bilaterally in the frontal lobes (Simister et al., 2003), or increased absolute concentration of Glx and decreased NAA in the thalamus (Bernasconi et al., 2003; Helms et al., 2006).
Seizure-associated MRS changes
MRS performed during or immediately after seizures can reveal seizure-induced metabolite changes including increased concentration of Lac on proton MRS and changes in tissue pH and energy-dependent metabolites on 31P MRS (Briellmann et al., 2005; Parmar et al., 2006). Post-ictal MRS in experimental dogs revealed increased concentration of Glx and lactate, and decreased concentration of Cr (Neppl et al., 2001).
Positron emission tomography and single-photon emission computed tomography
Molecular imaging with ictal and interictal positron emission tomography (PET) and sin-gle-photon emission computed tomography (SPECT) has clinical and research applications. PET and SPECT are used to identify the epileptogenic foci in the pre-operative assessment of patients with intractable focal epilepsy (La Fougère et al., 2009). Functional imaging with PET and SPECT reflects seizure-related changes of glucose uptake and metabolism (fluorine-18 fludeoxyglucose (18F)FDG-PET), neuroreceptor status (neuroreceptor PET) and cerebral perfusion (SPECT). The glucose analogue fluorine-18 fludeoxyglucose is an indirect marker of neuronal activity and allows absolute quantification of cerebral glucose metabolism. An epileptogenic focus will typically appear as an area of hypometabolism on interictal (18F)FDG-PET and an area of hypermetabolism on ictal (18F)FDG-PET. Ictal (18F)FDG-PET is not routinely performed due to logistic difficulties due mainly to the short half-life of the 18F label (La Fougère et al., 2009). The sensitivity of interictal (18F)FDG-PET in localizing the epileptogenic foci has been reported as 70–85% in patients with temporal lobe epilepsy (TLE) and 30–60% in patients with extra-TLE (Drzezga et al., 1999; Juhasz et al., 2001). (18F)FDG-PET correctly lateralized the seizure focus in 95% of MRI positive, 69% of MRI equivocal and 84% of MRI negative patients with temporal lobe epilepsy (Gok et al., 2012). Neuroreceptor PET of GABAA, serotonin (5-HT1A), opioid and dopamine receptors allows investigation of the role of these neurotransmitters in seizure generation and propagation, and helps developing new treatment approaches (La Fougère et al., 2009). PET with 18F-trans-4-fluoro-N-2(4-(2-methoxyphenyl) piperazin-1-yl)ethyl-N-(2-pyridyl) cyclohexanecarboxamide (18FFCWAY), a selective 5HT1A receptor antagonist, may help to localize mesial temporal epileptogenic foci in MRI-negative TLE patients (Liew et al., 2009; Theodore et al., 2012). In addition, PET has been used to evaluate the function of the efflux transporter P-glycoprotein (P-gp) at the blood-brain barrier (BBB) by means of specific P-gp PET tracers (Syvänen and Eriksson, 2013). P-gp restricts substrate compounds from entering the brain and may thus contribute to pharmacoresistance observed in certain patients with pharmacoresistant epilepsy.
SPECT entails use of 99mTc-HMPAO (technetium-99m hexamethylpropylene amine oxime) or 99mTc-ECD (technetium-99m-ethyl cysteinate diethylester) as substrate to assess regional cerebral blood flow changes during both the ictal and interictal periods. The epileptic focus will typically manifest as an area of hypoperfusion in the interictal stage and hyperperfusion in the ictal stage. Ictal SPECT is superior to interictal SPECT for identification of the location or the lateralization of epileptic foci in patients with TLE, with sensitivities between 73 and 97% for ictal SPECT and only 50% for interictal SPECT (Devous et al., 1998; Spanaki et al., 1999; Weil et al., 2001; Zaknun et al., 2008). However, in patients with extra-TLE, sensitivity of ictal SPECT is only 66% (Weil et al., 2001; Lee et al., 2008). Digital subtraction of ictal and interictal SPECT coregistered to MRI (referred to as SISCOM, subtraction ictal SPECT coregistered to MRI) is a multimodal imaging modality, which combines the structural and functional imaging information to improve the ability to detect and define the extent of epileptogenic foci in patients with normal conventional MRI scans (von Oertzen et al., 2011).
Functional MRI
Functional MRI (fMRI) has been used for the pre-operative assessment of people with intractable focal epilepsy and in neurophysiologic research (Sunaert and Yousry, 2001). fMRI relies on blood oxygenation level-dependent (BOLD) contrast: a change in the signal strength of brain water protons produced by the paramagnetic effects of venous blood deoxyhaemoglobin (Ogawa et al., 1993). BOLD fMRI measures the change in deoxyhaemoglobin concentration resulting from changes in cerebral blood flow, cerebral blood volume and cerebral metabolic rate of oxygen. fMRI permits the localization of brain areas associated with various motor, sensory and cognitive tasks and to assess their anatomic relationship to the seizure focus. This is of particular relevance in the planning of neurosurgical resection in people. Functional MRI can also be used to assess functional changes associated with the electroencephalographic onset of seizures. Sequential analysis of data generated by simultaneous EEG and fMRI recording allowed identification of cortical areas involved in the generation of interictal epileptiform activity in patients with pharmacoresistant extratemporal epilepsy (Donaire et al., 2013).
Cerebrospinal fluid analysis
The main role of cerebrospinal fluid (CSF) analysis in animals presenting with a history of seizures is to investigate structural brain disease, particularly inflammatory/infectious CNS disorders. Abnormalities of routine CSF analysis (red blood cell (RBC) count, white blood cell (WBC) count, cytological analysis and protein concentration) are relatively sensitive indicators of CNS disease; however, they are rarely specific for individual aetiologies. Occasionally, bacteria, Ehrlichia morulae, fungi, protozoa, parasites, Prototheca spp., canine distemper virus inclusion bodies, or neoplastic cells (e.g. lymphoblasts) may be identified on microscopic examination of CSF. More commonly, the main diagnostic value of CSF analysis is to narrow the differential diagnosis list of structural brain diseases causing seizures. This can be achieved by interpreting results of CSF routine analysis in the context of the animal’s signalment, medical history, neuroanatomic diagnosis and results of other diagnostic investigations including minimum database and imaging. Additional investigations for specific pathogens can be performed on CSF (see infectious disease testing) and may result in an aetiologic diagnosis. Quantification of organic acids, amino acids and other metabolites in CSF as well as urine and serum can assist in attaining a diagnosis of inborn errors of metabolism (Sargan, 2007).
CSF is an ultrafiltrate of plasma that is produced predominantly by the choroid plexi within the brain’s ventricular system and partly by the ependymal lining of the ventricular system, the pia-glial membrane and blood vessels in the pia-arachnoid. CSF flows through the ventricular system, into the sub-arachnoid space and it is subsequently absorbed predominantly through the arachnoid villi into the venous system (Tipold, 2003; Di Terlizzi and Platt, 2006). CSF has several functions, including regulation of intracranial pressure, regulation of the chemical environment of the CNS and intracerebral transport of biologically active substances including neurotransmitters and neuropeptides, the concentration of which may be altered in epileptic dogs (see Chapter 1) (Podell and Hadjiconstantinou, 1997, 1999).
Fig. 10.36. CSF is obtained by cistern magna puncture with the animal in lateral recumbence during general anaesthesia. The head and neck are near the table’s edge. The head is held flexed at a right angle to the cervical spine and the muzzle is held so that its long axis is parallel to the table. The landmark is a triangle composed from the occipital protuberance (A) and the most prominent point of the wings of the atlas (B). The puncture follows in the middle of this triangle one-third to one-half of the way caudal to the occipital protuberance, depending on breed anatomy. The needle is positioned on the midline, perpendicular to the atlanto-occipital space, and it is advanced slowly. The stylet has been removed before penetrating the dura mater in order to observe CSF flow into the hub of the needle as soon as the subarachnoid space is entered.
In animals investigated for seizures, CSF is usually collected from the cerebellomedullary cistern (or cisterna magna). The risks associated with this procedure include: iatrogenic brainstem trauma, transtentorial or foramen magnum herniation of neural tissue in animals with increased intracranial pressure, haemorrhage, general anaesthesia and iatrogenic CNS infection (Cook and DeNicola, 1988; Tipold, 2003; Di Terlizzi and Platt, 2006; Luján Feliu-Pascual et al., 2008). Contraindications to CSF collection include clinical suspicion (e.g. progressive loss of consciousness, papilloedema, changes in pupil size and response to light, decerebrate posture) or MRI signs of increased intracranial pressure, underlying coagulopathy or active intracranial haemorrhage, high anaesthetic risk, as well as atlanto-axial sub-luxation or other cervical vertebral instability and Chiari-like malformation in case of cerebellomedullary puncture. In animals with suspected structural epilepsy, performing MRI of the brain prior to CSF collection minimizes the risk associated with this procedure. If MRI of the brain is normal and inflammatory/infectious CNS disorders are suspected, CSF should be collected as inflammatory CSF changes may present despite the lack of MRI abnormalities (Lamb et al., 2005; Bohn et al., 2006).
CSF collection is performed with the animal in lateral recumbence during general anaesthesia. The head is held flexed at a right angle to the cervical spine without compromising airway patency and ventilation. The nose is slightly elevated in order to position the long axis of the muzzle parallel to the table. After sterile preparation of the region and confirmation of adequate depth of anaesthesia, the cerebellomedullary cistern puncture is performed between the occipital bone and the atlas using a 22-gauge, 1.5-inch spinal needle with a stylet (Fig. 10.36). A 2.5 inch spinal needle may be needed in large or giant breed dogs. A 22- or 25-gauge hypodermic needle can be used for very small-sized dogs and cats. The operator wears sterile gloves and carefully inserts the needle perpendicularly to the cranial cervical spine. The stylet can be removed before penetrating the dura mater in order to observe CSF flow into the hub of the needle as soon as the subarachnoid space is entered. Sometimes increased resistance to the needle advancement is perceived during penetration of the atlanto-occipital membrane and dura mater. The operator needs to be aware that CSF may not flow into the hub of the needle in animals with certain diseases resulting in increased CSF viscosity
(e.g. FIP) or obliteration of the cerebellomedullary cistern and therefore the needle should not be advanced too far. The CSF is allowed to drip into a sterile plain tube for routine analysis, antibody titres and microbial culture and in an EDTA tube for PCRs. The quantity of CSF, which can be safely obtained in the majority of animals, is 1 ml/5 kg of the animal’s body weight (Tipold, 2003). Usually 0.5 to 1 ml of CSF is adequate for routine analysis by most laboratories. An aliquot of CSF is saved for further analysis, such as antibody titres, PCRs, aerobic and anaerobic bacterial culture and susceptibility testing, or fungal culture, if indicated. Cell count and cytology preparations must be performed as soon as possible and ideally within 1 h of collection to minimize cellular degradation. If the sample is kept refrigerated at 4°C, a delay in processing of up to 8 h should not affect diagnostic interpretation. If a delay of more than 8 h is anticipated, hetastarch (1:1, volume basis) or serum (11% autologous serum or 20% fetal calf serum, volume basis) can be added to the CSF to preserve the refrigerated sample for at least 48 h (Bienzle et al., 2000; Fry et al., 2006). The addition of serum increases the protein concentration and therefore a separate CSF aliquot without serum should be kept refrigerated for protein quantification. Hetastarch does not affect the protein concentration of the CSF sample. The total number of cells present in CSF is determined by use of a cell counting chamber, such as a Fuchs-Rosenthal chamber. Cytological examination is done following concentration of the cells by cytocentrifugation or sedimentation chamber and staining (e.g. DiffQuick, Papanicolaou). Precise CSF protein quantification is done spectrophotometrically in commercial laboratories.
Normal CSF is clear and colourless, contains no RBC (unless there has been iatrogenic blood contamination) and 0 to 5 WBC/ml. Total protein concentration is less than 25 mg/dl in cerebellomedullary CSF samples and less than 45 mg/dl in lumbar cistern CSF samples. Reference intervals for CSF total protein concentration can vary with the laboratory and testing method used. Cytological examination of normal canine CSF reveals predominantly lymphocytes (60–70%) and monocytes (30–40%) and sometimes also a few segmented non-degenerate neutrophils (<1–9%) and rare erythrocytes (unless there has been iatrogenic blood contamination) (Di Terlizzi and Platt, 2006). In normal feline CSF monocytes are more numerous (69–100%) than lymphocytes (0–27%), and other cell types are rare as in dogs (Rand et al., 1990). Occasionally, meningeal lining cells, choroid plexus cells and ependymal cells can be found in normal CSF (Di Terlizzi and Platt, 2006).
An increase in the number of WBC in the CSF is referred to as pleocytosis, which may be classified as mild (6–50 WBC/ml), moderate (51–200 WBC/ml), or marked (>200 WBC/ml) and is further defined by the predominant cell type in the sample as mononuclear, neutrophilic, eosinophilic or mixed (Tipold, 2003).
The type of pleocytosis may be suggestive of a particular aetiology or disease group:
• Mononuclear pleocytosis (Plate 29):
• mild to moderate in viral meningoencephalitis (other than FIP) and breed-specific necrotizing encephalitis;
• moderate to marked in rickettsial and protozoal infections, granulomatous meningoencephalomyelitis (GME), bacterial meningitis following antibiotic treatment, chronic phase of steroid responsive meningitisarteritis (SRMA), CNS lymphoma.
• Neutrophilic pleocytosis (Plate 30):
• mild to moderate in brain necrosis associated with severe seizure activity, neoplasia, haemorrhage, infarction, granulocytic ehrlichiosis or anaplasmosis;
• moderate to marked in acute or subacute bacterial infections, feline infectious peritonitis (FIP), SRMA.
necrosis;
• moderate to marked in GME, rickettsial, protozoal, mycotic and prototheca infections, chronic FIP, chronic bacterial infections, chronic phase of SRMA, and rarely CNS histiocytosis.
The degree of CSF pleocytosis reflects the degree of leptomeningeal and/or ependymal involvement in the disease process, but it does not necessarily correlate with CNS disease severity or prognosis.
CSF total protein concentration is frequently increased in many disease processes that cause CSF pleocytosis. However, CSF protein concentration may be elevated without a concurrent increase in CSF nucleated cell count due to alterations in the blood-brain barrier or intrathecal globulin production. This phenomenon is referred to as albuminocytological dissociation or protein-cytological dissociation and can occur following recent seizures as well as in certain viral, anomalous, neoplastic, traumatic, vascular and degenerative CNS disorders (Di Terlizzi and Platt, 2009).
Iatrogenic contamination of the CSF sample with blood at the time of collection may complicate interpretation as it can falsely increase protein concentration, RBC count, WBC count and relative percentage of neutrophils and eosinophils. Blood contamination (>500 RBC/ml) in canine CSF containing less than 5 WBC/ml was significantly associated with an increase in protein concentration, percentage of neutrophils and presence of eosinophils (Doyle and Solano-Gallego, 2009). Activated macrophages and reactive lymphocytes were not affected by blood contamination, suggesting that the presence of these cells may be a more specific indicator of CNS disease in animals with normal CSF WBC count and blood contamination (Doyle and Solano-Gallego, 2009). In contrast, another study in dogs found no significant association between RBC count (mean 1285/ml, range 0–13,230/ml) and WBC count or protein concentrations in CSF from clinically normal or neurologically affected dogs (Hurtt and Smith, 1997). In feline CSF, a RBC count >30/ml can affect the total and differential WBC count (Rand et al., 1990). Formulas to correct nucleated cell count and total protein concentration for the effect of blood contamination in CSF are considered inaccurate (Sweeney and Russell, 2000).
Pathological haemorrhage within the CSF may be associated with infectious, inflammatory, traumatic and neoplastic disease. Erythrophagia, haemosiderophages or haematoid crystals on microscopic examination suggest pathological haemorrhage.
CSF may be normal, despite cerebral pathology, when CNS disease does not involve the leptomeninges or the ependymal lining of the ventricular system or if the animal has been treated with anti-inflammatory medications (particularly corticosteroids) prior to CSF collection.
Seizure-associated CSF changes
CSF pleocytosis (generally mild) may also occur as a result of seizure activity (Prokesh et al., 1983; Devinsky et al., 1988; Gonçalves et al., 2010). Proposed pathogenesis of this phenomenon involves a transient breakdown of the blood-brain barrier and release of chemotactic substances into the CSF during seizures (Schmidley and Simon, 1981; Edwards et al., 1983). A study in idiopathic epileptic dogs identified an association between CSF WBC count and time interval between the last seizure and the collection of the CSF. The longer the time interval, the lower the CSF WBC count. No association was found between CSF protein concentration and time of CSF collection as well as between the occurrence of cluster seizures and either the CSF WBC or protein concentration (Gonçalves et al., 2010).
Infectious disease testing
Infectious disease testing is performed when encephalitis or meningoencepahlitis is the suspected underlying aetiology of the seizures. Specific pathogens may be suspected based on the animal’s signalment, history (including geographic regions in which the animal has lived, travelled and currently lives), clinical signs and results of other diagnostic investigations including minimum database, thoracic and abdominal imaging, CSF routine analysis and MRI. Results of any infectious disease testing should be interpreted in the context of this information.
Immunologic testing measures antibodies against a specific pathogen in serum, CSF or both (e.g. canine distemper virus, Toxoplasma gondii, Neospora caninum, Ehrlichia spp., Anaplasma spp. and Rickettsia rickettsii) or antigen concentrations in serum, CSF or urine (e.g. Cryptococcus, Aspergillus and Blastomyces dermatitidis) (Berthelin et al., 1994; Spector et al., 2008; Garcia et al., 2012). The presence of antibodies indicates exposure and immune response towards an infectious agent, but does not confirm active infection. In addition, antibodies may result from previous vaccination against the agent or cross-reactivity with antibodies produced against another agent (e.g. feline infectious peritonitis virus (FIPV) and feline enteric corona-virus (FECV)). Evaluating both IgM and IgG, which reflect acute and chronic infection, respectively, performing serial antibody titres, or calculating the antibody index (IgG or antigen-specific) may help to support the suspected aetiologic diagnosis (Nghiem and Schatzberg, 2010). Recent or active infection is suggested by the presence of IgM, an increasing antibody titre over 2 to 3 weeks, or seroconversion (negative antibody result on the first test and positive antibody result 2–3 weeks later) (Lappin, 2009).
The IgG antibody index can help detecting intrathecal IgG synthesis suggesting the presence of the infectious agent in the CNS, and can be calculated with this formula:
IgGCSF/IgG serum
AlbuminCSF/albumin serum The main limitations of the IgG antibody index are that quantification of CSF albumin is not always readily available, and the index is not disease specific, it may be normal in the acute stage of canine distemper virus (CDV) and possibly other encephalitidies and may be elevated with lymphoid tumours or meningiomas (Tipold, 2003). An antigen-specific antibody index (ratio or coefficient) is more accurate than the IgG antibody index in detecting intrathecal antibody production (Furr, 2002) and can be calculated as follows:
(suspected disease-specific IgG in CSF/ suspected disease-specific IgG in serum) × (CAV or CPV specific IgG in CSF/CAV or CPV specific IgG in serum)
where CAV = canine adenovirus, CPV = canine parvovirus.
A ratio >1 indicates intrathecal IgG production against the suspected infectious disease. Ideally the same methodology should be used to quantify IgG for suspected and control infectious agents.
Antibody assays can be negative in per-acute infections if humoral immune responses have not had time to develop and in immune-compromised animals that may be unable to mount a humoral immune response.
Detection of antigen concentration in body fluids or tissue is more specific and may be more sensitive than antibody titres for certain pathogens (e.g. Blastomyces dermatitidis) (Spector et al., 2008), however it relies on the presence of the microorganism in the evaluated sample.
Molecular diagnostic testing such as conventional polymerase chain reaction (PCR), reverse transcriptase (RT)-PCR and other permutations of conventional PCR can detect and exponentially amplify small amounts of the infectious agent’s nucleic acids (DNA or RNA) in biological fluids or tissues (Nghiem and Schatzberg, 2010; Veir and Lappin, 2010). PCR or RT-PCR can be performed on CSF for various infectious agents including CDV, FIPV, Borna virus, Toxoplasma gondii, Neospora caninum, Rickettsia spp., Ehrlichia spp. and Streptococcus (Messer et al., 2008; Nghiem and Scatzberg, 2010; Veir and Lappin, 2010). PCR testing can give false positive (e.g. re-amplification of previously positive PCR reactions) or false negatives (e.g. the pathogen’s nucleic acids are present in the CNS parenchyma but are absent or present at undetectable levels in the CSF sample; the causative pathogen is no longer present in the CNS; inappropriate sample handling, particularly for RNA viruses) (Nghiem and Schatzberg, 2010). Results of PCR testing must be interpreted in conjunction with results of immunological testing, as well as other diagnostic investigations and the animal’s clinical presentation. The presence of infectious agent’s nucleic acid in a sample indicates that the infectious agent’s nucleic material exists in the host, but it does not demonstrate that the infectious agent is alive, capable of replication or actually causing clinical signs in the host (Veir and Lappin, 2010). Advances in PCR technology can increase the sensitivity and specificity of this test.
Aerobic and anaerobic bacterial culture (in blood, urine and CSF) and antimicrobial susceptibility testing and fungal culture in CSF or other body specimens should be performed any time these infectious agents are suspected, however sensitivity is generally low.
Genetic testing
Genetic tests can allow relatively inexpensive diagnosis of certain disorders that can result in seizure activity (Table 10.8). All disorders listed in Table 10.8 are autosomal recessive and result in interictal neurological abnormalities, with the exception of mildly to moderately affected Lagotto Romagnolo dogs which may have only seizures. It is likely that genetic testing will have a greater role in the diagnosis of canine epilepsy in the future as new genetic mutations are discovered.
Electroencephalography
Electroencephalography (EEG) is the recording of spontaneous electrical activity generated by pyramidal neurons in the cerebral cortex. EEG is widely used in investigation of seizure disorders in people and has been instrumental to seizure and epilepsy classification and treatment. Its use in veterinary medicine
Table 10.8. Genetic mutations resulting in seizures disorders (Penderis et al., 2007; Ekenstedt et al., 2012; Farias et al., 2012).
Gene mutated Canine breed Disorder Age at onset
| EPM2B | Miniature wirehaired |
| dachshunds | |
| LGI2 | Lagotto Romagnolo |
| L-2-hydroxyglutaric | Staffordshire bull terriers, |
| dehydrogenase | Yorkshire terriers |
| CLN8 | English setter |
| CLN5 | Border collie |
| CTSD | American bulldog |
| TPP1 | Dachshund |
| PPT1 | Dachshund |
| ARSG | American Staffordshire |
| terrier | |
| CLN6 | Australian shepherd |
| ATP13A2 | Tibetan terrier |
Lafora disease (myoclonic epilepsy) Benign familial juvenile epilepsy
L-2-hydroxyglutaric aciduria
Neuronal ceroid lipofuscinosis Neuronal ceroid lipofuscinosis Neuronal ceroid lipofuscinosis Neuronal ceroid lipofuscinosis Neuronal ceroid lipofuscinosis Neuronal ceroid lipofuscinosis Neuronal ceroid lipofuscinosis Neuronal ceroid lipofuscinosis
6–9 years 5–9 weeks (spontaneous
resolution by 8–13
weeks) 4–8 months 1–2 years >15 months <2 years 9 months <9 months 3–5 years <2 years Adult
is increasing, although still limited by patient cooperation, length of recording, equipment availability and expertise. The clinical applications of EEG in the seizure patient include: detection of epileptiform activity (e.g. spikes, sharp waves, spike and slow wave complexes, multiple spike complexes), localization of the epileptogenic focus, identification of focal or diffuse abnormalities of cerebral function (e.g. localized slowing of the background activity with focal structural cerebral disease or generalized periodic sharp waves with triphasic morphology with metabolic encephalopathies) and monitoring response to AEMs (Poncelet and Poma, 2013). Further information on EEG is presented in Chapter 11.
References
Arrol, L., Penderis, J., Garosi, L., Cripps, P., Gutierrez-Quintana, R. and Goncalves, R. (2012) Aetiology and long-term outcome of juvenile epilepsy in 136 dogs. Veterinary Record 170, 335.
Ashburner, J. and Friston, K.J. (2000) Voxel-based morphometry – the methods. Neuroimage 11, 805–821.
Aykut-Bingol, C., Tekin, S., Ince, D. and Aktan, S. (1997) Reversible MRI lesions after seizures. Seizure 6(3), 237–239.
Bailey, C.S. and Kitchell, R.L. (1987) Cutaneous sensory testing in the dog. Journal of Veterinary Internal Medicine 1, 128–135.
Basser, P.J. and Pierpaoli, C. (1996) Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. Journal of Magnetic Resonances. Series B 111, 209–219.
Bernasconi, A., Bernasconi, N., Natsume, J., Antel, S.B., Andermann, F. and Arnold, D.L. (2003) Magnetic resonance spectroscopy and imaging of the thalamus in idiopathic generalized epilepsy. Brain 126, 2447–2454.
Berthelin, C.F., Legendre, A.M. and Bailey, C.S. (1994) Cryptococcosis of the nervous system in dogs, part 2: diagnosis, treatment, monitoring, and prognosis. Progress in Veterinary Neurology 5, 136–146.
Bhatti, S.F., Vanhaesebrouck, A.E., Van Soens, I., Martlé, V.A., Polis, I.E., Rusbridge, C. and Van Ham, L.M. (2011) Myokymia and neuromyotonia in 37 Jack Russell terriers. Veterinary Journal 189(3), 284–288.
Bienzle, D., McDonnell, J.J. and Stanton, J.B. (2000) Analysis of cerebrospinal fluid from dogs and cats after 24 and 48 h of storage. Journal of the American Veterinary Medical Association 216, 1761–1764.
Bohn, A.A., Wills, T.B., West, C.L., Tucker, R.L. and Bagley, R.S. (2006) Cerebrospinal fluid analysis and magnetic resonance imaging in the diagnosis of neurologic disease in dogs: a retrospective study. Veterinary Clinical Pathology 35, 315–320.
Briellmann, R.S., Wellard, R.M. and Jackson, G.D. (2005) Seizure-associated abnormalities in epilepsy: evidence from MR imaging. Epilepsia 46(5), 760–766.
Bush, W.W., Barr, C.S., Darrin, E.W., Shofer, F.S., Vite, C.H. and Steinberg, S.A. (2002) Results of cerebrospinal fluid analysis, neurologic examination findings, and age at the onset of seizures as predictors for results of magnetic resonance imaging of the brain in dogs examined because of seizures: 115 cases (1992–2000). Journal of American Veterinary Medical Association 220(6), 781–784.
Cervera, V., Mai, W., Vite, C.H., Johnson, V., Dayrell-Hart, B. and Seiler, G.S. (2011) Comparative magnetic resonance imaging findings between gliomas and presumed cerebrovascular accidents in dogs. Veterinary Radiology and Ultrasound 52, 33–40.
Chahboune, H., Mishra, A.M., DeSalvo, M.N., Staib, L.H., Purcaro, M., Scheinost, D., Papademetris, X., Fyson, S.J., Lorincz, M.L., Crunelli, V., Hyder, F. and Blumenfeld, H. (2009) DTI abnormalities in anterior corpus callosum of rats with spike-wave epilepsy. Neuroimage 47, 459–466.
Chan, S., Chin, S.S., Kartha, K. et al. (1996) Reversible signal abnormalities in the hippocampus and neocortex after prolonged seizures. American Journal of Neuroradiology 17(9), 1725–1731.
Chang, Y., Mellor, D.J. and Anderson, T.J. (2006) Idiopathic epilepsy in dogs: owners’ perspectives on management with phenobarbitone and/or potassium bromide. Journal of Small Animal Practice 47(10), 574–581.
Chernov, M.F., Ochiai, T., Ono, Y., Muragaki, Y., Yamane, F., Taira, T., Maruyama, T., Tanaka, M., Iseki, H., Kubo, O., Okada, Y., Hori, T. and Takakura, K. (2009) Role of proton magnetic resonance spectroscopy in preoperative evaluation of patients with mesial temporal lobe epilepsy. Journal of the Neurological Sciences 285(1–2), 212–219.
Cizinauskas, S., Fatzer, R., Schenkel, M., Gandini, G. and Jaggy, A. (2003) Can idiopathic epilepsy be confirmed in cats? Journal of Veterinary Internal Medicine 17, 246.
Cole, A.J. (2004) Status epilepticus and periictal imaging. Epilepsia 45(Suppl. 4), 72–77.
Concha, L., Beaulieu, C., Collins, D.L. and Gross, D.W. (2009) White-matter diffusion abnormalities in temporal lobe epilepsy with and without mesial temporal sclerosis. Journal of Neurology, Neurosurgery and Psychiatry 80, 312–319.
Concha, L., Livy, D.J., Beaulieu, C., Wheatley, B.M. and Gross, D.W. (2010) In vivo diffusion tensor imaging and histopathology of the fimbria-fornix in temporal lobe epilepsy. Journal of Neuroscience 30, 996–1002.
Connelly, A., Van Paesschen, W., Porter, D.A., Johnson, C.L., Duncan, J.S. and Gadian, D.G. (1998) Proton magnetic resonance spectroscopy in MRI-negative temporal lobe epilepsy. Neurology 51(1), 61–66.
Cook, J.R. and DeNicola, D.B. (1988) Cerebrospinal fluid. Veterinary Clinics of North America Small Animal Practice 18, 475–499.
Cooper, J.J., Young, B.D., Hoffman, A., Bratton, G., Hicks, D.G., Tidwell, A. and Levine, J.M. (2010) Intracranial magnetic resonance imaging artifacts and pseudolesions in dogs and cats. Veterinary Radiology and Ultrasound 51(6), 587–595.
De Lahunta, A. and Glass, E. (2009) The neurological examination. In: Veterinary Neuroanatomy and Clinical Neurology. Elsevier-Saunders, pp. 487–501.
Devinsky, O., Nadi, S., Theodore, W.H. and Porter, R.J. (1998) Cerebrospinal fluid pleocytosis following simple, complex partial, and generalized tonic-clonic seizures. Annals of Neurology 23(4), 402–403.
Devous Sr, M.D., Thisted, R.A., Morgan, G.F., Leroy, R.F. and Rowe, C.C. (1998) SPECT brain imaging in epilepsy: a meta-analysis. Journal of Nuclear Medicine 39, 285–293.
Di Terlizzi, R. and Platt, S. (2006) The function, composition and analysis of cerebrospinal fluid in companion animals: part I - function and composition. Veterinary Journal 172(3), 422–431.
Di Terlizzi, R. and Platt, S.R. (2009) The function, composition and analysis of cerebrospinal fluid in companion animals: part II - analysis. Veterinary Journal 180(1), 15–32.
Donaire, A., Capdevila, A., Carreño, M., Setoain, X., Rumià, J., Aparicio, J., Campistol, J., Padilla, N., Sanmartí, F., Vernet, O., Pintor, L., Boget, T., Ortells, J. and Bargalló, N. (2013) Identifying the cortical substrates of interictal epileptiform activity in patients with extratemporal epilepsy: An EEG-fMRI sequential analysis and FDG-PET study. Epilepsia 30. doi: 10.1111/epi.12091.
Doyle, C. and Solano-Gallego, L. (2009) Cytologic interpretation of canine cerebrospinal fluid samples with low total nucleated cell concentration, with and without blood contamination. Veterinary Clinical Pathology 38(3), 392–396.
Drzezga, A., Arnold, S., Minoshima, S., Noachtar, S., Szecsi, J., Winkler, P., Römer, W., Tatsch, K., Weber, W. and Bartenstein, P. (1999) 18F-FDG PET studies in patients with extratemporal and temporal epilepsy: evaluation of an observer-independent analysis. Journal of Nuclear Medicine 40, 737–746.
Edwards, R., Schmidley, J.W. and Simon, R.P. (1983) How often does a CSF pleocytosis follow generalized convulsions? Annals of Neurology 13, 460–462.
Ekenstedt, K.J., Patterson, E.E. and Mickelson, J.R. (2012) Canine epilepsy genetics. Mammalian Genome 23(1–2), 28–39.
Farias, F.H., Zeng, R., Johnson, G.S., Shelton, G.D., Paquette, D. and O’Brien, D.P. (2012) A L2HGDH initiator methionine codon mutation in a Yorkshire terrier with L-2-hydroxyglutaric aciduria. BMC Veterinary Research 26(8), 124.
Fisher, R.S., van Emde Boas, W., Blume, W., Elger, C., Genton, P., Lee, P. and Engel, J. Jr (2005) Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46, 470–472.
Foster, E.S., Carrillo, J.M. and Patnaik, A. (1988) Clinical signs of tumours affecting the rostral cerebrum in 43 dogs. Journal of Veterinary Internal Medicine 2, 71–74.
Freeman, R.A. and Raskin, R.E. (2001) Cytology of the central nervous system. In: Raskin, R.E. and Meyer, D.J. (eds) Atlas of Canine and Feline Cytology. Saunders, Philadelphia, Pennsylvania, pp. 325–365.
Fry, M.M., Vernau, W., Kass, P.H. and Vernau, K.M. (2006) Effects of time, initial composition, and stabilizing agents on the results of canine cerebrospinal fluid analysis. Veterinary Clinical Pathology 35, 72–77.
Furr, M. (2002) Antigen-specific antibodies in cerebrospinal fluid after intramuscular injection of ovalbumin in horses. Journal of Veterinary Internal Medicine 16, 588–592.
Garcia, R.S., Wheat, L.J., Cook, A.K., Kirsch, E.J. and Sykes, J.E. (2012) Sensitivity and specificity of a blood and urine galactomannan antigen assay for diagnosis of systemic aspergillosis in dogs. Journal of Veterinary Internal Medicine 26(4), 911–919.
Garosi, L. and Lowrie, M. (2013a) The neurological examination. In: Platt, S. and Olby, N. (eds) BSAVA Manual of Canine and Feline Neurology, 4th edn. BSAVA, pp. 1–24.
Garosi, L. and Lowrie, M. (2013b) Lesion localisation and differential diagnoses. In: Platt, S. and Olby, N. (eds) BSAVA Manual of Canine and Feline Neurology, 4th edn. BSAVA, pp. 25–35.
Goffin, K., Dedeurwaerdere, S., Van Laere, K. and Van Paesschen, W. (2008) Neuronuclear assessment of patients with epilepsy. Seminars in Nuclear Medicine 38(4), 227–239.
Gok, B., Jallo, G., Hayeri, R., Wahl, R. and Aygun, N. (2012) The evaluation of FDG-PET imaging for epileptogenic focus localization in patients with MRI positive and MRI negative temporal lobe epilepsy. Neuroradiology. [Epub ahead of print]
Gonçalves, R., Anderson, T.J., Innocent, G. and Penderis, J. (2010) Effect of seizures on cerebrospinal fluid analysis in dogs with idiopathic epilepsy. Veterinary Record 166, 497–498.
Hecht, S. and Adams, W.H. (2010) MRI of Brain Disease in Veterinary Patients Part 1: basic principles and congenital brain Disorders. The Veterinary Clinics of North America Small Animal Practice 40, 21–38.
Helms, G., Ciumas, C., Kyaga, S. and Savic, I. (2006) Increased thalamus levels of glutamate and glutamine (Glx) in patients with idiopathic generalised epilepsy. Journal Neurology Neurosurgery and Psychiatry 77, 489–494.
Hiremath, G.K. and Najm, I.M. (2007) Magnetic resonance spectroscopy in animal models of epilepsy. Epilepsia 48(Suppl. 4), 47–55.
Huang, Y.C., Weng, H.H., Tsai, Y.T., Huang, Y.C., Hsiao, M.C., Wu, C.Y., Lin, Y.H., Hsu, H.L. and Lee, J.D. (2009) Periictal magnetic resonance imaging in status epilepticus. Epilepsy Research 86(1), 72–81.
Hurtt, A.E. and Smith, M.O. (1997) Effects of iatrogenic blood contamination on results of cerebrospinal fluid analysis in clinically normal dogs and dogs with neurologic disease. Journal of American Veterinary Medical Association 211(7), 866–867.
Juhász, C., Chugani, D.C., Muzik, O., Shah, A., Shah, J., Watson, C., Canady, A. and Chugani, H.T. (2001) Relationship of flumazenil and glucose PET abnormalities to neocortical epilepsy surgery outcome. Neurology 56, 1650–1658.
Jung, M.A., Nahm, S.S., Lee, M.S., Lee, I.H., Lee, A.R., Jang, D.P., Kim, Y.B., Cho, Z.H. and Eom, K.D. (2010) Canine hippocampal formation composited into three-dimensional structure using MPRAGE. Journal of Veterinary Medicine Science 72, 853–860.
Kim, J.A., Chung, J.I., Yoon, P.H., Kim, D.I., Chung, T.S., Kim, E.J. and Jeong, E.K. (2001) Transient MR signal changes in patients with generalized tonicoclonic seizure or status epilepticus: periictal diffusion-weighted imaging. American Journal Neuroradiology 22(6), 1149–1160.
Kimiwada, T., Juhasz, C., Makki, M., Muzik, O., Chugani, D.C., Asano, E. and Chugani, H.T. (2006) Hippocampal and thalamic diffusion abnormalities in children with temporal lobe epilepsy. Epilepsia 47, 167–175.
Kube, S.A., Vernau, K.M. and LeCouteur, R.A. (2006) Dyskinesia associated with oral phenobarbital administration in a dog. Journal of Veterinary Internal Medicine 20, 1238–1240.
Kuwabara, T., Hasegawa, D., Kobayashi, M. et al. (2010) Clinical magnetic resonance volumetry of the hippo-campus in 58 epileptic dogs. Veterinary Radiology and Ultrasound 51, 485–490.
La Fougère, C., Rominger, A., Förster, S., Geisler, J. and Bartenstein, P. (2009) PET and SPECT in epilepsy: a critical review. Epilepsy & Behaviour 15(1), 50–55.
Lamb, C.R., Croson, P.J., Cappello, R. and Cherubini, G.B. (2005) Magnetic resonance imaging findings in 25 dogs with inflammatory cerebrospinal fluid. Veterinary Radiology & Ultrasound 46(1), 17–22.
Lappin, M.R. (2009) Infectious disease diagnostic assays. Topics in Companion Animal Medicine 24(4), 199–208.
Lee, J.J., Lee, S.K., Lee, S.Y., Park, K.I., Kim, D.W., Lee, D.S., Chung, C.K. and Nam, H.W. (2008) Frontal lobe epilepsy: clinical characteristics, surgical outcomes and diagnostic modalities. Seizure 17, 514–523.
Liew, C.J., Lim, Y.M., Bonwetsch, R., Shamim, S., Sato, S., Reeves-Tyer, P., Herscovitch, P., Dustin, I., Bagic, A., Giovacchini, G. and Theodore, W.H. (2009) 18F-FCWAY and 18F-FDG PET in MRI-negative temporal lobe epilepsy. Epilepsia 50(2), 234–239.
Lorenz, M.D., Coates, J.D. and Kent, M. (2011) Neurologic History, Neuroanatomy, and Neurologic Examination. In: Handbook of Veterinary Neurology, 5th edn. Elsevier-Saunders, pp. 15–36.
Luján Feliu-Pascual, A., Garosi, L., Dennis, R. and Platt, S. (2008) Iatrogenic brainstem injury during cerebellomedullary cistern puncture. Veterinary Radiology & Ultrasound 49(5), 467–471.
MacKillop, E. (2011) Magnetic resonance imaging of intracranial malformations in dogs and cats. Journal of Veterinary Radiology and Ultrasound 52(1)(Suppl. 1), S42–51.
Mellema, L.M., Koblik, P.D., Kortz, G.D., LeCouter, R., Chechowitz, M.A. and Dickinson, P.J. (1999) Reversible magnetic resonance imaging abnormalities in dogs following seizures. Veterinary Radiology and Ultrasound 40(6), 588–595.
Messer, J.S., Wagner, S.O., Baumwart, R.D. et al. (2008) A case of canine streptococcal meningoencephalitis diagnosed using universal bacterial polymerase chain reaction assay. Journal of American Animal Hospital Association 44(4), 205–209.
Milne, M.E., Anderson, G.A., Chow, K.E., O’Brien, T.J., Moffat, B.A. and Long, S.N. (2013). Description of technique and lower reference limit for magnetic resonance imaging of hippocampal volumetry in dogs. American Journal of Veterinary Research 74(2), 224–231. doi:10.2460/ajvr.74.2.224
Mirrione, M.M. and Tsirka, S.E. (2011) Neuroimaging in Animal Seizure Models with (18) FDG-PET. Epilepsy Research and Treatment 369295, 8 pp.
Mishra, A.M., Bai, H., Gribizis, A. and Blumenfeld, H. (2011) Neuroimaging biomarkers of epileptogenesis. Neuroscience Letters 27, 497(3), 194–204.
Najm, I.M., Wang, Y., Shedid, D., Lüders, H.O., Ng, T.C. and Comair, Y.G. (1998) MRS metabolic markers of seizures and seizure-induced neuronal damage. Epilepsia 39(3), 244–250.
Neppl, R., Nguyen, C.M., Bowen, W., Al-Saadi, T., Pallagi, J., Morris, G., Mueller, W., Johnson, R., Prost, R. and Rand, S.D. (2001) In vivo detection of postictal perturbations of cerebral metabolism by use of proton MR spectroscopy: preliminary results in a canine model of prolonged generalized seizures. American Journal of Neuroradiology 22(10), 1933–1943.
Nghiem, P.P. and Schatzberg, S.J. (2010) Conventional and molecular diagnostic testing for the acute neurologic patient. Journal of Veterinary Emergency and Critical Care 20(1), 46–61.
Ogawa, S., Menon, R.S., Tank, D.W., Kim, S.G., Merkle, H., Ellermann, J.M. and Ugurbil, K. (1993) Functional brain mapping by blood oxygenation level-dependent contrast magnetic resonance imaging. A comparison of signal characteristics with a biophysical model. Biophysical Journal 64(3), 803–812.
Pákozdy, A., Leschnik, M., Tichy, A.G. and Thalhammer, J.G. (2008) Retrospective clinical comparison of idiopathic versus symptomatic epilepsy in 240 dogs with seizures. Acta Veterinaria Hungarica 56(4), 471–483.
Pákozdy, A., Thaller, D., Gumpenberger, M., Leschnik, M., Galler, A., Shibly, S. and Klang, A. (2013) Concurrent bilateral temporal lobe pathology and unilateral oligodendroglioma in a dog with status epilepticus. Journal of Small Animal Practice 54(2), 112–113.
Parmar, H., Lim, S.H., Tan, N.C. and Lim, C.C. (2006) Acute symptomatic seizures and hippocampus damage: DWI and MRS findings. Neurology 13, 66(11), 1732–1735.
Penderis, J., Calvin, J., Abramson, C., Jakobs, C., Pettitt, L., Binns, M.M., Verhoeven, N.M., O’Driscoll, E., Platt, S.R. and Mellersh, C.S. (2007) L-2-hydroxyglutaric aciduria: characterisation of the molecular defect in a spontaneous canine model. Journal of Medical Genetics 44, 334–340.
Podell, M. and Hadjiconstantinou, M. (1997) Cerebrospinal fluid gammaaminobutyric acid and glutamate values in dogs with epilepsy. American Journal of Veterinary Research 58, 451–456.
Podell, M. and Hadjiconstantinou, M. (1999) Low concentrations of cerebrospinal fluid GABA correlate to a reduced response to phenobarbital therapy in primary canine epilepsy. Journal of Veterinary Internal Medicine 13, 89–94.
Podell, M., Fenner, W.R. and Powers, J.D. (1995) Seizure classification in dogs from a nonreferral-based population. Journal of the American Veterinary Medical Association 206, 1721–1728.
Poncelet, L. and Poma, R. (2013) Electrophysiology. In: Platt, S. and Olby, N. (eds) BSAVA Manual of Canine and Feline Neurology, 4th edn. BSAVA, pp. 72–76.
Powell, H.W., Parker, G.J., Alexander, D.C., Symms, M.R., Boulby, P.A., Barker, G.J., Thompson, P.J., Koepp, M.J. and Duncan, J.S. (2001) In vivo detection of postictal perturbations of cerebral metabolism by use of proton MR spectroscopy: preliminary results in a canine model of prolonged generalized seizures. American Journal Neuroradiology 22, 1933–1943.
Powell, H.W., Parker, G.J., Alexander, D.C., Symms, M.R., Boulby, P.A., Wheeler-Kingshott, C.A., Barker, G.J., Koepp, M.J. and Duncan, J.S. (2005) MR tractography predicts visual field defects following temporal lobe resection. Neurology 65, 596–599.
Powell, H.W., Parker, G.J., Alexander, D.C., Symms, M.R., Boulby, P.A., Barker, G.J., Thompson, P.J., Koepp, M.J. and Duncan, J.S. (2008) Imaging language pathways predicts postoperative naming deficits. Journal of Neurology, Neurosurgery and Psychiatry 79, 327–330.
Prokesch, R.C., Rimland, D., Petrini, J.L. and Fein, A.B. (1983) Cerebrospinal fluid pleocytosis after seizures. Southern Medical Journal 76, 322–327.
Quesnel, A.D., Parent, J.M., McDonell, W., Percy, D. and Lumsden, J.H. (1997) Diagnostic evaluation of cats with seizure disorders: 30 cases (1991–1993). Journal of the American Veterinary Medical Association 210, 65–71.
Rand, J.S., Parent, J., Jacobs, R. and Percy, D. (1990) Reference intervals for feline cerebrospinal fluid: Cell counts and cytologic features. American Journal of Veterinary Research 51, 1044–1048.
Robertson, I. (2011) Optimal magnetic resonance imaging of the brain. Veterinary Radiology and Ultrasound 52(1)(Suppl. 1), S15–S22.
Ródenas, S., Pumarola, M., Gaitero, L., Zamora, A. and Añor, S. (2011) Magnetic resonance imaging findings in 40 dogs with histologically confirmed intracranial tumours. Veterinary Journal 187, 85–91.
Ross, B. and Bluml, S. (2001) Magnetic resonance spectroscopy of the human brain. The Anatomical Record 265, 54–84.
Sargan, D.R. (2007) Inherited metabolic disease in companion animals: prospects for their diagnosis and elimination in the next decade. Veterinary Journal 174(2), 223–224.
Schmidley, J.W. and Simon, R.P. (1981) Postictal pleocytosis. Annals of Neurology 9, 81–84.
Schriefl, S., Steinberg, T.A., Matiasek, K., Ossig, A., Fenske, N. and Fischer, A. (2008) Etiologic classification of seizures, signalment, clinical signs, and outcome in cats with seizure disorders: 91 cases (2000–2004). Journal of the American Veterinary Medical Association 233, 1591–1597.
Schwartz, M., Muñana, K.R. and Nettifee-Osborne, J. (2013) Assessment of the prevalence and clinical features of cryptogenic epilepsy in dogs: 45 cases (2003-2011). Journal of the American Veterinary Medical Association 242(5), 651–657.
Seidenberg, M., Kelly, K.G., Parrish, J., Geary, E., Dow, C., Rutecki, P. and Hermann, B. (2005) Ipsilateral and contralateral MRI volumetric abnormalities in chronic unilateral temporal lobe epilepsy and their clinical correlates. Epilepsia 46, 420–430.
Shihab, N., Bowen, J. and Volk, H.A. (2011) Behavioral changes in dogs associated with the development of idiopathic epilepsy. Epilepsy and Behaviour 21(2), 160–167.
Simister, R.J., McLean, M.A., Barker, G.J. and Duncan, J.S. (2003) Proton MRS reveals frontal lobe metabolite abnormalities in idiopathic generalized epilepsy. Neurology 14, 61(7), 897–902.
Simister, R.J., McLean, M.A., Salmenpera, T.M., Barker, G.J. and Duncan, J.S. (2008) The effect of epileptic seizures on proton MRS visible neurochemical concentrations. Epilepsy Research 81(1), 36–43.
Simister, R.J., McLean, M.A., Barker, G.J. and Duncan, J.S. (2009) Proton MR spectroscopy of metabolite concentrations in temporal lobe epilepsy and effect of temporal lobe resection. Epilepsy Research 83(2–3), 168–176.
Smith, M.O., Turrel, J.M., Bailey, C.S. and Gain, G.R. (1989) Neurologic abnormalities as the predominant signs of neoplasia of the nasal cavity in dogs and cats: seven cases (1973–1986). Journal of American Veterinary Medical Association 195, 242–245.
Smith, P.M., Talbot, C.E. and Jeffery, N.D. (2008) Findings on low-field cranial MR images in epileptic dogs that lack interictal neurological deficits. Veterinary Journal 176(3), 320–325.
Smith, R.A. and Lang, D.G. (2000) Rapid determination of ethylene glycol and glycolic acid in biological fluids. Veterinary and Human Toxicology 42(6), 358–360.
Spanaki, M.V., Spencer, S.S., Corsi, M., MacMullan, J., Seibyl, J. and Zubal, I.G. (1999) Sensitivity and specificity of quantitative difference SPECT analysis in seizure localization. Journal of Nuclear Medicine 40, 730–736.
Spector, D., Legendre, A.M., Wheat, J., Bemis, D., Rohrbach, B., Taboada, J. and Durkin, M. (2008) Antigen and antibody testing for the diagnosis of blastomycosis in dogs. Journal of Veterinary Internal Medicine 22(4), 839–843.
Sunaert, S. and Yousry, T.A. (2001) Clinical applications of functional magnetic resonance imaging. NeuroImaging Clinics of North America 11, 221–236.
Sweeney, C.R. and Russell, G.E. (2000) Differences in total protein concentration, nucleated cell count, and red blood cell count among sequential samples of cerebrospinal fluid from horses. Journal of American Veterinary Medical Association 217(1), 54–57.
Syvänen, S. and Eriksson, J. (2013) Advances in PET Imaging of P-Glycoprotein Function at the Blood-Brain Barrier. ACS Chemical Neuroscience 20, 4(2), 225–237
Szabó, C.A., Lancaster, J.L., Lee, S., Xiong, J.H., Cook, C., Mayes, B.N. and Fox, P.T. (2006) MR imaging volumetry of subcortical structures and cerebellar hemispheres in temporal lobe epilepsy. American Journal of Neuroradiology 227, 2155–2160.
Theodore, W.H., Martinez, A.R., Khan, O.I., Liew, C.J., Auh, S., Dustin, I.M., Heiss, J. and Sato, S. (2012) PET of serotonin 1A receptors and cerebral glucose metabolism for temporal lobectomy. Journal of Nuclear Medicine 53(9), 1375–1382.
Thomas, W.B. (2010) Evaluation of veterinary patients with brain disease. The Veterinary Clinics of North America Small Animal Practice 40(1), 1–19.
Tipold, A. (2003) Cerebrospinal Fluid. In: Braund, K.G. (ed.) Clinical Neurology in Small Animals - Localization, Diagnosis and Treatment . International Veterinary Information Service (www.ivis.org), Ithaca, New York.
Van Hee, P., Neels, H., De Doncker, M., Vrydags, N., Schatteman, K., Uyttenbroeck, W., Hamers, N., Himpe, D. and Lambert, W. (2004) Analysis of gamma-hydroxybutyric acid, DL-lactic acid, glycolic acid, ethylene glycol and other glycols in body fluids by a direct injection gas chromatography-mass spectrometry assay for wide use. Clinical Chemistry and Laboratory Medicine 42(11), 1341–1345.
Veir, J.K. and Lappin, M.R. (2010) Molecular diagnostic assays for infectious diseases in cats. The Veterinary Clinics of North America Small Animal Practice 40(6), 1189–1200.
Von Oertzen, T.J., Mormann, F., Urbach, H., Reichmann, K., Koenig, R., Clusmann, H., Biersack, H.J. and Elger, C.E. (2011) Prospective use of subtraction ictal SPECT coregistered to MRI (SISCOM) in presurgical evaluation of epilepsy. Epilepsia 52(12), 2239–2248.
Vullo, T., Deo-Narine, V., Stallmeyer, M.J., Gomez, D.G. and Cahill, P.T. (1996) Quantitation of normal canine hippocampus formation volume: correlation of MRI with gross histology. Magnetic Resonance Imaging 14, 657–662.
Weil, S., Noachtar, S., Arnold, S., Yousry, T.A., Winkler, P.A. and Tatsch, K. (2001) Ictal ECD-SPECT differentiates between temporal and extratemporal epilepsy: confirmation by excellent postoperative seizure control. Nuclear Medicine Communications 22, 233–237.
Wisner, E.R., Dickinson, P.J. and Higgins, R.J. (2011) Magnetic resonance imaging features of canine intracranial neoplasia. Veterinary Radiology and Ultrasound 52(1)(Suppl. 1), 52–61.
Wolff, C.A., Holmes, S.P., Young, B.D., Chen, A.V., Kent, M., Platt, S.R., Savage, M.Y., Schatzberg, S.J., Fosgate, G.T. and Levine, J.M. (2012) Magnetic resonance imaging for the differentiation of neoplastic, inflammatory, and cerebrovascular brain disease in dogs. Journal of Veterinary Internal Medicine 26(3), 589–597.
Young, B.D., Levine, J.M., Porter, B.F., Chen-Allen, A.V., Rossmeisl, J.H., Platt, S.R., Kent, M., Fosgate, G.T. and Schatzberg, S.J. (2011) Magnetic resonance imaging features of intracranial astrocytomas and oligodendrogliomas in dogs. Veterinary Radiology and Ultrasound 52(2), 132–141.
Zaknun, J.J., Bal, C., Maes, A., Tepmongkol, S., Vazquez, S., Dupont, P. and Dondi, M. (2008) Comparative analysis of MR imaging, ictal SPECT and EEG in temporal lobe epilepsy: a prospective IAEA multi-center study. European Journal of Nuclear Medicine and Molecular Imaging 35, 107–115.
Zhu, H. and Barker, P.B. (2011) MR Spectroscopy and Spectroscopic Imaging of the Brain. Methods in Molecular Biology 711, 203–226.
11 Introduction to Electroencephalography
Fiona James
Department of Clinical Studies, Ontario Veterinary College, University of Guelph, Ontario, Canada
Principles of Electroencephalography
Electroencephalography refers both to the study and the technique of recording electrical activity from the brain via electrodes placed upon the head (Chatrian et al., 1974). Thus, the electroencephalogram (EEG) is a neurological ‘tool’ for the investigation of cortical brain function. This tool has been developed since the late 19th century when preliminary work showed that the living brain generated electrical impulses that could be measured with electrodes and a galvanometer (Caton, 1875). It has become a useful test in the diagnosis of epilepsy, particularly in human medicine where it is required in the definition of epileptic electroclinical syndromes (Berg et al., 2010).
Electroclinical syndromes are defined by a typical age of onset, specific EEG characteristics, seizure semiology and other features such that this complex of clinical features together form a distinctive, recognizable clinical disorder. The specific diagnosis of an electroclinical syndrome can become the focus of diagnostic investigations (including genetic testing) and treatment trials. EEG has also found a place in the monitoring of cortical function in human patients in the intensive care unit, as well as in the field of anaesthesia to monitor depth of sedation and anaesthesia. Technologically, the field of EEG instrumentation has developed to encompass synchronous video EEG (v-EEG), capability for continuous EEG recording (cEEG) and, most recently, wireless/remote recording.
A complete understanding of the EEG requires knowledge of the underlying electrophysiological basis and anatomic substrates of the various expected waveforms. Artefacts may be either physiologic or non-physiologic. Physiologic artefacts are those arising from the patient’s body, e.g. the electrocardiogram (ECG). Various physical factors also contribute to the quality of the EEG, including EEG instrumentation, selection of electrode types, pattern of electrode placement or montage for viewing EEG results and methods of patient handling, including manual versus pharmaceutical restraint. Choice of pharmaceutical may affect the expected EEG waveforms, and the implications are yet to be fully understood.
With computerization and digital EEG available, there are now two techniques for EEG analysis: visual and automated. Software allows power analysis of an EEG, breaking down the waveform into its constituent frequencies, as well as topographic analysis to localize the focus of abnormalities. Algorithms are available for ‘live’ analysis, triggering an alarm if certain threshold conditions are met,
e.g. the appearance of epileptiform discharges, or a change in anaesthetic depth. The original
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
approach of simple visual inspection requires time and experience; in order to interpret the EEG, one must first recognize what is normal. Expected waveforms will depend on the age and vigilance state of the patient, as well as the influence (if any) of administered pharmaceuticals.
Electrophysiological basis
There are essentially two types of cells that compose the grey and white matter of the central nervous system: neurons and glial cells. The former are specialized for communication and the latter for neurosupportive functions and are interspersed between the neurons throughout the brain. Neurons have two types of cellular processes: dendrites that conduct signals towards the cell body (soma) and axons that conduct signals away from the soma. The cell bodies of the neurons lie in layers (laminae) in the grey matter, typically oriented such that their dendrites are more superficial than their somas, with their axis perpendicular to the surface of the cerebral cortex (Fig. 11.1) (King, 1987).
Neurons communicate with each other via synapses with incoming axons. The neuronal cell membrane derives its electrical potential from the creation of ion concentration gradients via ion channels in its membrane. Incoming signals at synapses result in small excitatory and inhibitory postsynaptic potentials (PSPs) that propagate over the dendrites and soma, interacting. Sufficient cumulative excitation above a certain threshold results in an action potential being generated. The PSP at the dendritic pole of the neuron causes a transient separation of charge (either positive or negative) via the changes in ionic concentrations, forming a dipole oriented along the neuronal axis (Holliday and Williams, 1999). Both extra- and intracellular ion currents flow along these dipoles due to the excellent conductivity of both media (Holliday and Williams, 1999). However, the discharge of a single neuron or nerve fibre, causing a single PSP is too small to be recorded from the surface of the head. Instead, the extracellular ion currents resulting from the simultaneous

I II
III
IV V
VI
Fig. 11.1. A three-dimensional representation of the laminae of the cerebral cortex. The cell bodies of the neurons are oriented perpendicular to the surface of the cortex, with their dendrites more superficial than their somas. The layers of the cortex are indicated with roman numerals, with layer I being the most superficial layer.
generation of PSPs in multiple neurons in a region produce extracellular field potentials that can be recorded by electrodes placed on the scalp, skull or surface of the cerebral cortex via volume conduction (Speckmann and Elger, 2005). Widespread interdigitating glial processes and interconnections appear to play an amplifying role in the genesis of the extracellular field potential (Amzica, 2002). As these current flows are oriented perpendicularly to the surface of the cortex along the axis of the neurons, electrodes on the head will record positive or negative potentials with respect to a distant reference electrode, dependent upon the direction of the current flow (Holliday and Williams, 1999; Speckmann and Elger, 2005). The amplitude of the recording at the scalp will result from the voltage of the cortical source, the area of the cortex
Electroencephalography
involved, the depth of the source and the synchronicity of the multiple neurons (Alarcon et al., 1994). Characteristics of the tissue and electrode dictate that the electrical activity recorded arises from the superficial cerebral cortex, from a depth of approximately 5 mm and a surface diameter of 1–2 cm (Holliday and Williams, 1999). Therefore, tissues deeper than the cortex, e.g. the thalamus, only influence the EEG indirectly.
The patterns of electrical activity of the brain vary rhythmically and can be characterized by their frequency and amplitude (Holliday and Williams, 1999). The fluctuations are confined to a range of frequencies between 0.5 and 50 Hz, with the majority of patterns occurring below 30 Hz (Holliday and Williams, 1999). The amplitudes of these waveforms vary between a few to several hundred microvolts (usually below 100 µV) (Holliday and Williams, 1999). Patterns in recordings appear when there is synchronized activity in the neuron population. This synchronized activity is typically dependent upon the level of excitation and is related to distinct behavioural states (Steriade et al., 1990). Simply put, high amplitude EEG waveforms with relatively slow frequencies are described as ‘synchronized’ activities (Steriade et al., 1990). Synchronicity occurs when networks of neurons discharge with the same frequency as a result of some form of interaction (Steriade et al., 1990). While some single neurons have demonstrated spontaneous rhythmic fluctuations in their membrane potentials (oscillations) in vitro, in vivo these neurons are subject to the influence of neuronal populations and networks (Steriade et al., 1990). Thus, the originators of synchronicity in the brain may be single neurons, groups of neurons acting in concert, or networks of neurons with feedback circuits (Steriade et al., 1990). These sources produce rhythmic variations in electrical activity whose synchronicity additively amplifies the electrical signal such that it may be measured by an EEG. Anatomic substrates for these neurons or networks include parts of the limbic system and the reticular thalamic nucleus, with multiple connections that distribute signal widely, or, in cases of focal EEG synchronicity (e.g. those seen during states of focused attention), circumscribed parts of the cerebral cortex (Steriade et al., 1990).
Conversely, blocking or attenuation of an EEG pattern implies a replacement of synchronized waveforms with lower amplitude and relatively faster waves (Chatrian et al., 1974; Steriade et al., 1990; Noachtar et al., 1999). This attenuation, as can be seen in alert subjects or during EEG-desynchronized paradoxical sleep (also known as rapid eye movement (REM) sleep), arises from blockage of synchronization by cholinergic projections arising from the brainstem and basal forebrain as part of the ascending reticular activating system (Steriade et al., 1990). Cholinergic input to thalamocortical neurons transforms them from the synchronous ‘burst’ firing mode of slow-wave sleep (also known as non-REM sleep, NREM sleep) to the more individual tonic repetitive firing seen in an alert state (Steriade et al., 1990). Thus, in awake and alert individuals, the EEG ‘background activity’ is the summation of the multiple individually active neurons manifesting as low amplitude, high frequency activity, with no specific patterns recognizable (Fig. 11.2).
Electroencephalography instrumentation
Originally analogue, and now digital in its latest incarnation, EEG instrumentation consists of the electrodes and the machine that records the ongoing electrical activity of the brain (Nuwer et al., 1998; Ebner et al., 1999). Each electrode is connected to the machine such that the signal of one electrode is subtracted from the other of a pair of electrodes, thus producing a measurement of the potential difference between the two electrodes, relative to the same reference electrode (Ebner et al., 1999). This suppresses voltage changes common to both electrodes, known as common mode rejection, a tactic used to reduce noise from, for example, a 60 Hz current as originates from the mains in North America (Ebner et al., 1999).
There is a polarity convention on displaying these recordings: an upward deflection indicates that input 1 is negative with respect to input 2, or that input 2 is positive
Pyramidal cells
Thalamic reticular cells
Thalamic relay cells
T-type Ca2+ channels
Depolarization Closed Hyperpolarization
Open Inactive
Fig. 11.2. The levels of circuitry involved in synchronicity when the thalamus is in burst-firing mode. Thalamic relay cells have T-type calcium channels in their membranes that oscillate between closed, open and inactive states, being open when the membrane is depolarized. Thalamic relay cells have excitatory input to layer IV pyramidal cells and thalamic reticular cells. Pyramidal cells, in turn, excite the thalamic reticular cells and thalamic relay cells. However, the thalamic reticular cells inhibit the thalamic relay cells.
with respect to input 1. Conversely, there is a downward deflection if input 1 is positive with respect to input 2, or if input 2 is negative with respect to input 1 (Ebner et al., 1999). This polarity convention has the confounding result that ‘up’ is ‘negative’ and ‘down’ is ‘positive’ on recording displays.
Adjustable high and low frequency filters (also known as ‘high-pass’ and ‘low-pass’ filters) offer the option of restricting the recording to the frequency band of interest. Over-restriction of the data can, obviously, result in the loss or distortion of information (Ebner et al., 1999). Similarly, a 60 Hz notch filter, while convenient for removing the North American 60 Hz mains artefact, may have the unintended consequence of ‘smoothing’ sharply contoured pathological waveforms (Nuwer et al., 1998; Ebner et al., 1999).
Patient and operator safety is enhanced by the grounded power cord, necessary in the current hospital setting where patients are often attached to multiple electrical devices simultaneous with conductive intravenous or bodily fluids (Ebner et al., 1999; Kamp et al., 2005).
Electrode types
Electrodes consist of a metal contact surface, a terminal end and flexible insulated wire in between. Intrinsic to an electrode’s ability to reproduce a waveform is its impedance, itself a function of electrode capacitance and resistance (Ebner et al., 1999; Reilly, 2005). EEG machines have a control panel provision for checking electrode impedance, which should be kept below 5 kW to avoid signal distortion or attenuation (Nuwer et al., 1998; Ebner et al., 1999; Reilly, 2005). The metal constituting the electrode will affect the accuracy of the waveform reproduction. In descending order of accuracy, electrode metals are: silver-silver chloride, copper, platinum, silver, gold and stainless steel (Klemm, 1965; Ebner et al., 1999).
For the routine clinical outpatient, minimally invasive electrode placement is recommended, either skin surface, subcutaneously or deep in the fascia, in combination with a form of physical or pharmaceutical restraint in order to the keep the electrodes in place (Croft, 1962; Fox, 1967a; Nuwer et al., 1993).
Electroencephalography
These superficial electrodes have an inherent disadvantage in that electrical signals from the brain are attenuated to approximately 1/7th of the intracortical level, and are often confused by artefacts from local muscle activity and electrode movements (Klemm, 1965). Large areas of cortex contribute to the waveforms recorded, allowing localization to • an approximately 10 mm area (Cuffin et al., 2001a, b). However, overall gross changes in the EEG can be detected with superficial electrodes that might be missed by studying more localized cortical regions at depth.
form of a bandage or medical tape, pull out easily and are invasive, albeit minimally. They suffer the same neuroimaging incompatibilities as the cup electrodes and are not recommended for long-term EEG recordings for humans (Engel et al., 1993; Mirsattari et al., 2005). Subdermal wire electrodes (SWEs) (Fig.11.3c) offer many advantages including rapidity and ease of application, decreased maintenance requirements, possibility of sterile placement and compatibility with both CT and MRI (Ives, 2005). However, they are easily removed and require a protective head covering. They are (minimally) invasive and may cause local bleeding in up to 40% of placements in humans (Ives, 2005). They also appear to be associated with greater amounts of non-physiologic artefact than the other two electrode types, most often an electrode pop, discussed in the artefact section below (James et al., 2011).

Comparisons of the SWEs with gold cup electrodes (GCEs) and SNEs have been performed (Ives, 2005; Young et al., 2006; James et al., 2011). For the short-term recording (<2 h), electrode type and sedation state do not appear to matter (James et al., 2011). For longer recordings, the clinician should be aware that persistent artefacts may develop in SWEs, but these may not be clinically significant for up to 8 h of recording (James et al., 2011). GCEs will require periodic maintenance beyond 24 h to reduce artefacts (Young et al., 2006).
Montage
The recording from a pair of electrodes is called a derivation (Holliday and Williams, 1999; Noachtar et al., 1999). A montage is an arrangement of derivations displayed simultaneously in an EEG record (Holliday and Williams, 1999; Noachtar et al., 1999). Montages are designed to allow accuracy and ease in identifying EEG events and their anatomical origins – two types typically used are bipolar and common reference montages (Holliday and Williams, 1999). With digital EEG, switching between montages is possible post hoc, known as ‘remontaging’ (Holliday and Williams, 2001).
• The reference montage (Fig. 11.4a) is formed from derivations wherein one electrode is used as the reference for all electrodes (Holliday and Williams, 1999; Noachtar et al., 1999). This reference electrode is ideally placed on an electrically inactive site on the body to minimize the likelihood of picking up the same EEG activity as the exploring electrode of the pair (Holliday and Williams, 1999; Noachtar et al., 1999). However, there is no such site on the body, and increasing distances from the head introduces artefact from other electrical sources in the body (e.g. the ECG), but its placement on the head means that the possibility should be considered that the reference electrode might be affected by large EEG events. In a reference montage, focal waveforms of interest are located based on their amplitude being larger than the background (Holliday and Williams, 1999).
• For a bipolar montage (Fig. 11.4b), the electrodes contributing to a derivation are both situated over active cerebral cortex, with no electrode being common to all derivations (Holliday and Williams, 1999). Usually, bipolar derivations are linked to form chains oriented either longitudinally and/or transversely across the head, such that each adjacent derivation has one electrode in common (Holliday and Williams, 1999; Noachtar et al., 1999). Arranged this way, an interesting waveform originating under one electrode will cause opposite deflections of the EEG tracing, an eye-catching feature termed an ‘instrumental phase reversal’ (Holliday and Williams, 1999).
Montages are constructed from electrodes placed on the head of the subject. For human EEG, the International Federation of Societies for Electroencephalography and Clinical Neurophysiology adopted a standard electrode placement protocol in 1957, known as the ten-twenty system (Jasper, 1957). The distance between external landmarks on the head is measured and the electrodes placed at 10 or 20% divisions of these measurements. Additionally, each electrode position was named topographically, e.g. Fp1 or Cz for Frontopolar 1 and vertex electrodes, respectively, with odd and even numbers referring to the left and right side of the head, respectively. This standard system facilitates the comparison of EEG records between facilities as well as the reporting of results in the literature. The standard system was devised by comparison of the electrode placement protocols then in use in the major EEG laboratories of the world, deriving the common factors and imposing a common nomenclature (Jasper, 1957).
In the history of veterinary EEG, the original approach was the two-channel machine with surgical needle electrodes inserted subcutaneously on the scalp and a ground electrode placed distantly on a dog under manual restraint (Croft, 1962). Focal anomalies were found by triangulation as the electrodes were moved around (Fox, 1967a). A more sophisticated awake EEG recording technique proposed an eight-channel EEG and subdermal pin-type

Electroencephalography
Fig. 11.4. (a) The background activity of a dog during REM sleep showing low-amplitude high-frequency activity with an eye movement to the left (large deflection in T3 channel); low-frequency filter (LF): 0.1 Hz, high-frequency filter (HF): 70 Hz, notch filter applied (60 Hz), horizontal scale 1 s, vertical scale 40 µV.
(b) Instrumental phase reversal during 8 Hz paroxysmal activity accompanying facial twitching centred on C4 in a dog; LF: 0.1 Hz, HF: 70 Hz, notch filter applied (60 Hz), horizontal scale 1 s, vertical scale 40 µV.
(a)
F3-REF
C3-REF
T3-REF
O1-REF
F4-REF
C4-REF
T4-REF
O2-REF
(b)
F3-F4 T3-C3 C3-C4 C4-T4 O1-O2 F3-C3 C3-O1 F3-T3 T3-01 F4-C4 C4-O2 F4-T4 T4-O2
REF F3 F4 T3C3 T4
C4 O1 O2
electrodes (Herin et al., 1968). The montage had electrode positions designated frontal, parietal, occipital and vertex, with the ground electrode placed subcutaneously just below the occiput (inion). The eight-channel recording montage is still reported, as is a five-channel montage (Holliday and Williams, 1998; Brauer et al., 2011, 2012). Others are moving towards modifications of the ten-twenty system, recording >8 channels with subjects under sedation or general anaesthesia (Bergamasco et al., 2003; Pellegrino and Sica, 2004; Williams et al., 2008). By 1988, 14 of 16 veterinary neurologists responding to a survey were placing electrodes according to the five-electrode pattern described by Redding and Knecht (Redding and Knecht, 1984; Steiss, 1988). However, in veterinary EEG, there is no standard electrode placement system, nor nomenclature. As with the ten-twenty system, a standard electrode placement system and nomenclature would be helpful in comparing results between laboratories and in the literature. Due to the varying shapes of animal heads, landmarks may have to differ between species and between breeds within a species in order to compare, for example, a dolichocephalic to a brachycephalic dog. Dissection and neuroimaging studies would be needed to confirm the anatomic correlate of electrode positions (Pellegrino and Sica, 2004; Lewis et al., 2011).
Patient handling
Along with electrode positioning, patient handling has been the subject of much discussion in veterinary EEG, not being as big a problem in human EEG. Its importance lies in the minimizing of movement artefacts in the EEG recording. Although recordings have been performed in awake unmedicated dogs, there is a risk that the movement artefact can obscure the recording, particularly for longer recordings (e.g. >2 h)(Croft, 1962; Redding and Knecht, 1984; Holliday and Williams, 1999; James et al., 2011). Three solutions have been suggested: adequate fixation of the electrodes as previously discussed, restriction of subject movement (physical or pharmaceutical restraint) or telemetry (Klemm, 1965). There are no standard methods for approaching patient handling, with various researchers choosing the method that seemed most practical for them.
Surgical approaches to restriction of subject movement have included de-afferentation of the thalamocortex, e.g. the encéphale isolé and the cerveau isolé (Klemm, 1965). This is not in common current usage due to obvious ethical and survival issues. Physical restraint methods range from slings, tie downs, form-fitting boxes or holders, to training (Klemm, 1965; Fox, 1967a). Manual restraint for EEG recording has been described, with assistants holding a dog by its leash and collar, or in lateral recumbency with eyes and ears covered and limbs held or taped (Croft, 1962; Fox, 1967a; Holliday et al., 1970). Covering the ears or using an audiometric testing chamber attenuates exposure to auditory stimuli (Holliday et al., 1970).
Various pharmaceuticals have been used to achieve chemical restraint in veterinary EEG, ranging from paralytics to sedatives to general anaesthetics (Klemm, 1965; Klemm and Mallo, 1966; Herin et al., 1968). Paralytics offer the benefit of removing muscle-generated artefacts from the recordings, however, the dogs must be intubated and maintained on a ventilator while conscious and alert, which presents ethical issues. Alternatively, several studies suggested that light propofol anaesthesia, alone or with paralytics, e.g. rocuronium, allows detection of both normal features and paroxysmal discharges in dogs while reducing muscle artefact (Accatino et al., 1997; Jaggy and Bernardini, 1998; Bergamasco et al., 2003; Akos et al., 2012).
Early interest in the effects of various sedative and anaesthetic drugs on the canine EEG has been developed into automated monitoring of depth of anaesthesia with, for example, propofol (Averill and de Lahunta, 1968; Kusters et al., 1998; Bergamasco et al., 2003; Lee et al., 2012). Anaesthetic drugs (amobarbital, pentobarbital, thiamylal and thiopental), when administered intravenously until the palpebral reflex can no longer be elicited, produced characteristic EEG changes that suggested that EEG levels of anaesthesia were sufficiently distinctive to allow monitoring of anaesthesia (Averill and de Lahunta, 1968). It has been asserted that anaesthesia caused less masking of EEG abnormalities than alert wakefulness (Klemm, 1968). Similarly, it has been stated that light sedation is optimal for recording EEGs from epileptic canine patients as abnormal waveforms are more likely to be encountered when the patient is drowsy, a state of vigilance unlikely to be encountered with an unsedated or anaesthetized patient (Holliday and Williams, 1998, 1999).
Xylazine has been investigated as a sedative (ranging from 0.5 to 1.5 mg/kg subcutaneously) for EEG recordings, with 1 mg/kg being the dose that achieved optimal restraint (Pellegrino and Sica, 2004). At lower doses, the dogs moved too much, creating muscle artefact. At higher doses, the dogs fell asleep, resulting in sleep patterns only. Recordings of EEG in neonate to adult beagle dogs under xylazine sedation showed unaltered, normal sleep patterns (Tourai et al., 1985). Xylazine thus showed promise as a sedative for EEG data collection with which a drowsy state may easily be achieved.
In dogs, sedation with chlorpromazine, promazine and propriopromazine at 1.1 mg/kg
Electroencephalography
showed minimal alterations to the control EEG patterns, suggesting that these sedatives, too, were acceptable for chemical restraint (Averill and de Lahunta, 1968). In contrast, an EEG study of 43 suspected primary epileptic dogs administered 2.2 mg/kg chlorpromazine by rapid intravenous bolus found that chlorpromazine altered the EEG pattern of epileptic dogs (Holliday et al., 1970). The study design had some flaws – there was a lack of blinding or placebo control – however, it appears that chlorpromazine may be helpful in activating the EEG of epileptic dogs. In a similar study, chlorpromazine (2 mg/kg), in combination with intermittent light stimulation (ILS), was found to increase the likelihood of synchronous EEG waveforms in epileptic beagles, but to minimal effect on normal beagles (Redman et al., 1973). More recently, sedation for EEG data collection was performed with acepromazine (0.1 mg/kg intravenously) in dogs, with the hypothesis that, if acepromazine had a provocative effect on the EEG of epileptic patients similar to that of chlorpromazine, this was advantageous as it increased the likelihood of recording anomalous waveforms (Holliday and Williams, 1998). However, retrospective studies of acepromazine administration in dogs with a history of seizures failed to find an association between the timing of the acepromazine dosage and the next occurrence of seizures (Tobias et al., 2006; McConnell et al., 2007). Contributing to the weakness of these studies were the retrospective nature, the lack of a control cohort, lack of blinding, lack of EEG recordings and the low number of dogs in the studies. Thus, there is evidence to support the use of chlorpromazine as a sedative for EEG data collection in that it does not alter the EEG in control dogs but may ‘activate’ the EEG in epileptic dogs. This utility has yet to be shown with acepromazine.
In cats, medetomidine hydrochloride (0.03–0.05 mg/kg intramuscularly), alone or in combination with butorphanol tartrate (0.01–0.02 mg/kg intravenously) has been used to successfully obtain EEG recordings in cats from 2 to 24 weeks of age (Wrzosek et al., 2009; Lewis et al., 2011). Alpha2-adrenoceptor agonists appear to result in high-voltage low-frequency background activity in the EEG for both dogs and cats, along with the manifestation of sleep spindles, k-complexes and vertex sharp transients, i.e. slow-wave sleep (Tourai et al., 1985; Farber et al., 1997; Wrzosek et al., 2009; Lewis et al., 2011). Other methods used with cats include inhalant anaesthesia, paralytics and sodium pentobarbital anaesthesia (Klemm, 1968; Hori et al., 1979; Bouyer et al., 1987; Wada et al., 1990).
Electroencephalography Interpretation
There are two techniques available for EEG analysis: visual and automated (computerized) analysis (Holliday and Williams, 1999). The original approach was simple visual inspection of the EEG recording; this requires time and experience. With the advent of computers and digital EEG, software algorithms may now provide ‘live’ frequency spectral analysis of an EEG during acquisition via fast Fourier transformation. This allows quantification of the amount (or ‘power’) of various frequency bands seen in the EEG recording over an epoch (a set period of time), resulting in a ‘frequency power spectrum’ (Holliday and Williams, 2003). Absolute band power and relative band power can be calculated and used to statistically compare between epochs of interest. The term ‘band’ refers to named subdivisions of the frequencies encountered on an EEG:
An ‘amplitude spectrum’ may also be calculated, giving a graphical presentation of the frequency analysis, making it easy to pick out the dominant frequency in an epoch. This numerical analysis of the frequency of the EEG is referred to as quantitative EEG, or q-EEG. Software analysis can also provide topographic mapping as to the focus of abnormal waveforms, or an alarm if certain threshold settings are reached (automated event detection). However, software findings still require human oversight for confirmation to rule out false positives or negatives, e.g. accidental inclusion of artefacts or exclusion of pathological waveforms (Holliday and Williams, 2001, 2003).
Regardless of automated or visual analysis technique, critical to the analysis of an EEG is the age and vigilance state of the patient (Niedermeyer, 2005). There are various stages of vigilance with EEG correlates: wakefulness, drowsiness, slow-wave sleep and paradoxical REM sleep, with division of non-REM sleep into several stages based on EEG characteristics (Fox, 1967b; Holliday and Williams, 1999; Williams et al., 2008). Understanding these normal EEG patterns associated with the states of vigilance and age allows the identification and distinguishing of normal from abnormal EEG activity and movement artefact from true EEG activity.
Against the background activity, certain rhythms may come to dominate and, therefore, to be identified.
limb zone of the primary somatosensory cortex. It is likely related to blockage of motor activity, also known as the ‘sensorimotor rhythm’, similar to that seen over the central sulcus in humans (Roth et al., 1967; Steriade et al., 1990). The 35–45 Hz peak occurs over the motor and parietal association cortices in cats watching an unattainable mouse (Bouyer et al., 1987; Steriade et al., 1990). It is speculated to represent maximal synaptic transmission related to focused motor programming (Steriade et al., 1990).
0.5 to 4 Hz (Noachtar et al., 1999). These slow waves are seen during natural sleep and originate from pyramidal neurons in the cortex (Ball et al., 1977; Steriade et al., 1990). Delta waves <1 Hz reflect oscillatory sequences of excitation and inhibition of cortical neurons that is cycled reflectively through the thalamus and back to the cortical networks and involves complex exchanges between neurons and surrounding glia modulated by the extra-cellular ionic environment (Amzica, 2002). Suppression of these waves upon arousal originates from the nucleus basalis in the basal forebrain via cholinergic projection (Steriade et al., 1990; Amzica, 2002) (Fig. 11.6).
During the early stages of quiet sleep,
transient ‘K-complexes’, ‘sleep spindle wave
forms’ and ‘vertex sharp transients’ (also known
Electroencephalography
as ‘V waves’) may be identified. They tend to be of highest amplitude over midline (Holliday and Williams, 1999). Sleep spindles are waxing and waning groups of rhythmic waves with frequencies between 7 and 14 Hz, grouped in sequences lasting for 1.5–2 s that recur with a frequency of 0.1–0.2 Hz, and that are spread widely over the cerebral cortex (Fox, 1967b; Steriade et al., 1990; Noachtar et al., 1999). K-complexes are a slow wave (large amplitude negative and positive deflection) often associated with sleep spindles (Holliday and Williams, 1999; Amzica, 2002) (Fig. 11.7). V waves are elicited by stimulation during drowsiness or early sleep, often from an auditory stimulus (Holliday and Williams, 1999).
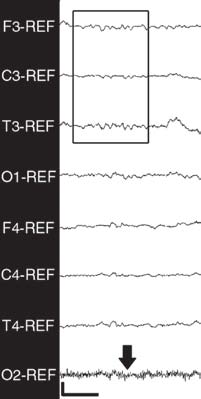

Vigilance state and the EEG
The vigilance state of the patient refers to the level of consciousness/arousal, helping to differentiate, for example, between an alpha rhythm in an awake individual and sleep spindles in a drowsy individual. Understanding the normal EEG patterns associated with the states of vigilance of a species enables recognition of abnormal EEG waveforms as well as investigation into disorders of sleep (Schubert et al., 2011). For such reasons, investigations in the normal sleep patterns of the human comprise a large body of literature. Such a body of literature is gradually being built in veterinary EEG. The baseline information provided by confirmed normal patterns allows comparisons to be made of the effects of drugs or disease. Such an atlas of reference EEG patterns is available for the dog and the cat, however this resource needs to be updated to cover the recordings taken from the multi-channel digital EEG machines now available (Redding and Knecht, 1984).
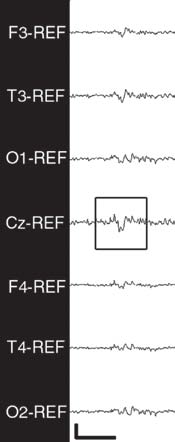
Four recognizable levels of vigilance have been described in the dog: (i) wakefulness;
(ii) drowsiness; (iii) slow wave quiet sleep; and (iv) REM (rapid eye movement) (Holliday and Williams, 1999). A fully aroused awake dog is one that responds to environmental stimuli with head and body movements, typically attempting to rise from recumbency. Unfortunately, this state is often accompanied by muscle artefact (intermittent or continuous high frequency EEG activity) or movement artefact (transient high amplitude activity). Either of these may obscure the background rhythm in at least one channel of the EEG of the awake dog (Holliday and Williams, 1999). Different physiological states of arousal may be correlated to specific frequency bands and can be subdivided as follows:
11.6 and 11.7).
4. In REM sleep, the dogs are as they are during slow-wave sleep, but the background rhythm resembles that of the awake state; eye movements may begin after 5–10 min of REM sleep, often accompanied by twitching of the paw, mouth or other body parts (Fox, 1967b; Holliday and Williams, 1999) (Fig 11.4a).
Age and the EEG
As an animal ages, the ‘normal’ EEG (in terms of frequency and voltage patterns) changes in a predictable way (Fox, 1967a, b, c; Redding and Knecht, 1984; Kellaway, 2003; Lewis et al., 2011). Knowing what EEG patterns to expect at which age aids the reviewer in determining what is abnormal.
In dogs from birth to 16 weeks of age, a marked increase in the overall amplitude was seen from 3 to 7 weeks of age that then declined thereafter (Charles and Fuller, 1956; Pampiglione, 1963; Fox 1967a, b, c). Basal frequency of the EEG recording showed a steady increase as pups matured, reaching that typical of adults by approximately 1 year of age (Pampiglione, 1963; Fox, 1967c). The typical sleep patterns of adults also gradually became more recognizable. In neonates, it was initially hard to distinguish sleep from awake states based on EEG patterns or behaviour. However, by 2 weeks of age this distinction was clear, along with slow-wave components of quiet sleep (Fox, 1967b).
A certain EEG pattern of bursts of 16–18 Hz activity with amplitude ranging from 50 to 150 µV, termed ‘neonatal spindles’, was described in pups up to 3 weeks of age, and was thought to be non-specific thalamocortical activity (Fox, 1967b). The occipital dominant alpha rhythm that appears when vision is occluded (eyes are closed) in an awake dog can be demonstrated in pups by 3 weeks of age (Fox, 1967b). Adult-style sleep spindles became apparent around 5 weeks of age (Fox, 1967b). Between 5 and 10 weeks of age, there was a further increase in the frequency of the fast components of the EEG recording and in the amplitude of slow waves (Fox, 1967b). From 12 to 16 weeks, the amplitude decreased again, but the frequencies continued to increase to those of adulthood; sleep patterns also matched those of the adult more closely (Fox, 1967b).
Cats also show significant modification in the EEG during maturation of the cerebral
Electroencephalography
cortex. For the first 2 weeks, the awake kitten showed periods of electrical discontinuity (intervals of very low amplitude) that is abolished by 4 weeks of age (Redding and Knecht, 1984; Lewis et al., 2011). The background activity amplitude was low in 2-week-old kittens, reaching a peak of approximately 17 µV by 6 to 8 weeks, before decreasing to reach almost adult levels between 20 and 24 weeks of age. Frequency of the background activity tended to be more irregular than in the adult cat. Absolute power for each frequency band also peaked around 6 to 8 weeks of age, and then diminished to a plateau from approximately 20 weeks onwards. Occasional transients have been reported in 2-week-old kittens (sharp waves and spike-and-slowwave discharges), as well as occasional 15 Hz spindles up to 4 weeks of age (Redding and Knecht, 1984; Lewis et al., 2011). Sleep spindles and K-complexes are apparent by 4 weeks and older, with sleep spindles visible in all leads with a frequency of 8–13 Hz (Redding and Knecht, 1984; Lewis et al., 2011).
Artefacts in the EEG
Artefacts may be divided into: (1) patient-origin, physiological artefact; and (2) external, non-physiologic artefact (Holliday and Williams, 1999; Klem, 2003).
and should be resolvable with replacement or adjustment of the electrode (Klem, 2003). This is because most electrode artefacts are due to either poorly attached electrodes, high resistance, broken wires or changes in the tissue– electrode interface, e.g. by drying of electrode jelly (Klem, 2003).
Faulty electrodes may develop abnormal capacitance or impedance.
Capacitance
Capacitance is an electrical engineering term for the amount of charge stored by a substance at a given voltage (Litt and Cranstoun, 2003). Biological elements with capacitance include cerebrospinal fluid, the bony skull and the scalp – all of these may alter the EEG signal, particularly at the electrode–scalp interface (Litt and Cranstoun, 2003). Fluctuations in electrode capacitance may result in variable charges building up on the electrode surface, causing transient and typically positive discharges, easily recognized because they occur on only one electrode and have a rapid rise with slow decay phase (Young et al., 2006). These are known as electrode ‘pops’ (Fig. 11.8).
Impedance
Impedance is an electrical engineering principle that describes the effects of the combination of electrical resistance, capacitance and inductive reactance (Litt and Cranstoun, 2003). There are several reasons why impedance is important in EEG. From a safety perspective, dangerously low impedances may allow high currents to pass through tissue, with obvious consequences. At the other end of the spectrum, high impedances, typically resulting from poorly affixed electrodes, disrupt the EEG’s amplifiers (in the circuit to magnify the signal from the brain) and thereby distort the resulting signal. Very high electrode impedances result in significant signal attenuation (Litt and Cranstoun, 2003). From a practical standpoint, this means that electrode impedances must be checked (and electrode attachment adjusted as necessary) before, during and after an EEG recording to ensure that they remain within the optimal range.
This feature is offered on most modern, digital EEG machines.
Pathological waveforms
Pathological waveforms may be categorized as interictal epileptiform abnormalities, ictal patterns associated with seizures and periodic epileptiform discharges (Chang and Drislane, 2007).
Epileptiform patterns are those distinctive waves or complexes that can be distinguished from the background and resemble those recorded in epileptic subjects (human or animal). They include spikes and sharp waves, either alone or accompanied by slow waves, and may occur singly or in bursts that last, at most, a few seconds. They refer only to interictal paroxysmal events, not to seizure patterns (Chatrian et al., 1974; Noachtar et al., 1999).
A paroxysm or paroxysmal discharge is defined as an EEG event with an abrupt onset, rapid attainment of a maximum and a sudden termination, and that can be distinguished from the background activity (Chatrian et al., 1974; Noachtar et al., 1999) (Fig. 11.9). Paroxysms are distinct from bursts, which are a group of waves that appear and disappear abruptly, being distinguished from the background by differences in frequency, form and/or amplitude; where ‘paroxysm’ is commonly used to refer to epileptiform or seizure patterns, ‘burst’ does not imply abnormality (Chatrian et al., 1974; Noachtar et al., 1999).
A spike is an EEG transient that is clearly distinguished from the background, with a pointed peak and a duration of approximately 20–70 ms (Chatrian et al., 1974; Noachtar et al., 1999). The main component usually has a negative polarity, and it may have variable amplitude. It is thought to be generated by the synchronous depolarization of the cortical neurons located within an approximately 6 cm2 area (Chang and Drislane, 2007).
A sharp wave is a transient that resembles a spike in every aspect except that it is of longer duration, 70–200 ms (Chatrian et al., 1974; Noachtar et al., 1999). Spikes and sharp waves are interictal epileptiform discharges that are generally considered to suggest an underlying tendency for epileptogenesis (Chang and Drislane, 2007).
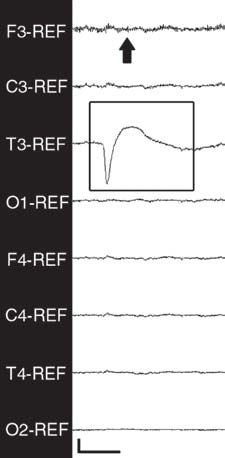

Electroencephalography
When combined with a slow wave, a spikeand-wave complex should be accompanied by a descriptor where appropriate, e.g. thephrase ‘multiple 4 Hz spike-and-wave complexes’ is to be preferred over ‘4 Hz polyspike-and-wave complex’ (Chatrian et al., 1974; Noachtar et al., 1999). Spike-and-slow-wave complexes are also strongly suggestive of an underlying epileptic disorder (Chang and Drislane, 2007). Multiple spike complexes (frequencies of ³10 Hz) may be observed with generalized seizure disorders in humans, and may have a myoclonic jerk associated with them (Chang and Drislane, 2007). When waves consist of two or more components that develop on either side of the baseline, they are referred to as polyphasic waves,
e.g. diphasic or triphasic waves (Chatrian et al., 1974; Noachtar et al., 1999). An example of a benign differential diagnosis for spikes and sharp waves is the vertex wave associated with sleep. Vertex waves tend to have a more symmetric morphology with phase reversal over the vertex (midline) channels, whereas a spike’s initial deflection is usually steeper than the return to baseline (Chang and Drislane, 2007). Spikes should also not be confused with electrode ‘pop’ artefacts – the initial deflection of the latter is often more steep and is followed by an exponential decay (Chang and Drislane, 2007).
‘Burst-suppression pattern’ – while officially recommended for use only to describe the EEG effects of some anaesthetic drugs at certain levels of anaesthesia, in practice this term is used to describe alternating stretches of bursts and low-voltage EEG, where the bursts may be spike(s) alone or mixed with delta or theta activity (Chatrian et al., 1974; Niedermeyer et al., 1999). The bursts typically last 100–1000 ms and the voltage ranges from 100 to 1000 µV. The suppression periods last longer than the bursts, and can be extremely long (minutes/ hours) in cases of brain death or prolonged barbiturate therapy. One should check for low voltage (5–20 µV) versus isoelectricity at the highest levels of amplification. For the most part, a burst-suppression pattern indicates either deep levels of anaesthesia (either with barbiturates or some inhalants, e.g. isoflurane or sedative overdoses) or severe (life-threatening) cerebral anoxia (Niedermeyer et al., 1999) (Fig. 11.10).

Ictal patterns are those recorded on an EEG during a clinical seizure; the ictal nature is suggested by the rhythmicity and evolution of the EEG pattern, especially for focal seizures where only a few channels may be affected (Chang and Drislane, 2007). A generalized tonic-clonic seizure is typically characterized by a rapid (>10 Hz) repetition of generalized spikes and spike complexes (Chang and Drislane, 2007). There is usually an increase in amplitude and concomitant decrease in frequency during the first 10–20 s that corresponds to the tonic phase observed clinically. The clonic phase is accompanied by bursts of generalized high-amplitude spike-and-slow-wave or multiple spike complexes that may be seen to coincide with the clonic jerks of the limbs. There is often a low-voltage background between these complexes. The burst frequency tapers off towards the end of the seizure. Post-ictally, the background is usually low-voltage, followed by generalized delta-frequency slowing. Physiologic (muscle) artefact often obscures these details during the seizure (Fig. 11.11).
Absence seizures in humans are similar to that reported in a Chihuahua (Chang and Drislane, 2007; Poma et al., 2010) (Fig. 11.12). A low frequency (3–5 Hz) burst of generalized spike-and-slow-wave may be recorded for up to a few seconds at a time, multiple times per day. The patient may stare unresponsively during the seizure, and it may be accompanied by minor automatisms.
Various neurologic diseases may result in periodic epileptiform discharges. Exploration of EEG in veterinary cases of head trauma, encephalitis, cerebrovascular accidents and intracranial space-occupying lesions demonstrate varying amounts of left-right asymmetry, low voltage (or runs of low voltage), 6–12 Hz slow waves, high-voltage slow waves, spikes and multiple spike-and-wave complexes (Croft, 1962, 1970a, b, 1971, 1972). EEG changes (spikes and spike-and-wave paroxysms) have been reported in a dog with post-status epilepticus neurologic dysfunction and necrosis of the amygdalae, parahippocampal gyri and extratemporal cortices (Hasegawa et al., 2005).
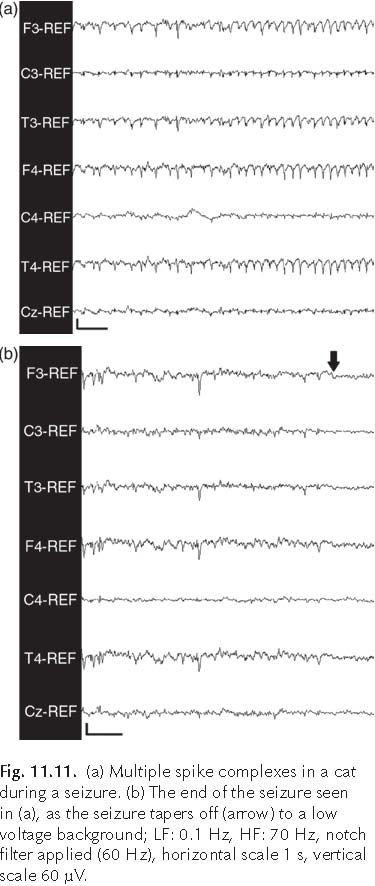
Clinical Applications of Electroencephalography
EEG represents an essential diagnostic test in human epilepsy allowing the assessment of many different aspects of cortical function in vivo. EEG recordings provide both evidence supporting diagnoses of human seizure disorders, as well as assistance in the classification of human epilepsy electroclinical syndromes (Sundaram et al., 1999; Berg et al., 2010).
Electroencephalography

Historically, the diagnosis of a seizure disorder depended on the recognition of the characteristic behavioural and physical features, or semiology, manifesting during a seizure event, thus allowing classification of seizure types. However, semiology alone can only suggest hemispheric lateralization of the seizure focus and does not provide sufficiently accurate localization of the epileptogenic site within the cerebral cortex (So, 2006). The specific diagnosis of an electroclinical epileptic syndrome allows research developments into genetic causes, diagnostic investigations and treatment trials.
There are several forms of EEG performed during epilepsy studies. The interictal routine EEG, a short (20–30 min) scalp EEG, is typically ordered on an outpatient basis in humans to diagnose specific electroclinical syndromes and to aid in localization of the seizure focus when considering epilepsy surgery for refractory epileptics (Fountain, 2006). In cases of single seizure events of unknown aetiology, routine EEGs may aid in the prediction of the possibility of seizure recurrence (Sundaram et al., 1999; Fountain, 2006). During the use of, and after the withdrawal of, anti-epileptic medications (AEMs) in human patients, routine EEGs may provide similarly useful prognostic information (Sundaram et al., 1999). Two activation procedures, hyperventilation and intermittent photic stimulation, are commonly employed for the human subject during routine EEG, i.e. to increase the likelihood of detecting an interictal epileptiform discharge (Chang and Drislane, 2007). Photic stimulation and hyperventilation may have an activating effect in dogs, with baseline non-responses reported in normal dogs, but do not appear to increase the likelihood of routine EEG finding interictal epileptiform activity in more than one-third of epileptic dogs (Holliday et al., 1970; Brauer et al., 2011, 2012). As drowsiness and sleep are also precipitants of epileptiform discharges, sedation may play a part in activating the veterinary EEG (discussed earlier) comparable to the human practice of performing EEGs after sleep deprivation (Chang and Drislane, 2007). Activation is important as only approximately 50% of humans with known epilepsy have interictal discharges on their first routine EEG (Chang and Drislane, 2007).
Another form of EEG study, long-term monitoring (LTM), using video-synchronized EEG (v-EEG), is indicated for clarifying the type of electroclinical syndrome, presurgical evaluation, differentiating epileptic from nonepileptic conditions, documentation of circadian variation in events (and effects of interventions), clarifying sleep-associated events, and monitoring in the intensive care unit for the treatment of convulsive and non-convulsive status epilepticus (SE) (Velis et al., 2007). Continuous electroencephalogram (cEEG, a form of LTM) recording for spontaneous electrical activity of the cerebral cortex is used in the intensive care monitoring of thalamocortical function in critically ill human patients, e.g. cases of status epilepticus, metabolic encephalopathy and coma, or to monitor response to AEM therapy (Young and Campbell, 1999; Scheuer, 2002; Kaplan, 2004, 2006). cEEG has been reported as an aid in the management of status epilepticus in dogs and cats (Raith et al., 2010). v-EEG is particularly helpful in humans with an imperfect ability to communicate and in differentiating epileptic from nonepileptic syndromes, hence its particular suitability for veterinary EEG (Cascino, 2002; Nordli, 2006). In a related area, v-EEG has been used for investigation into sleep disorders of dogs, where it enabled confirmation of the diagnosis of REM sleep behaviour disorder (Schubert et al., 2011). The technology is now available for wireless outpatient v-EEG LTM (also known as ambulatory LTM), a tool that might well have been tailored to veterinary specifications.
In certain human epilepsy cases that are refractory to AEM therapy, identification of the epileptogenic focus location within the cerebral cortex allows surgical intervention. This takes the form of either subpial cortical transection (for areas of eloquent, or language processing, cortex that still retain function) or focal cortical excision (for cortex that is not eloquent, and the removal of which will not be associated with significant neurologic deficits), up to and including lobectomy or hemispherectomy (Blume and Schomer, 1988; Blume, 1997). This potential therapeutic option for refractory canine epileptics has yet to be fully explored.
The advent of digital EEG and advances in software capabilities have expanded the role of EEG in these situations; the development of seizure-prediction algorithms has been a vibrant area of study since the 1970s (Castellaro et al., 2002; Litt and Echauz, 2002; Mormann et al., 2007; Lesser and Webber, 2008). One might envision pocket or implanted EEG monitors that indicate when to initiate therapy in advance of an impending seizure. Other uses of automated EEG analysis include monitoring the depth of anaesthesia in surgical patients and monitoring brain function in critical care patients (Kusters et al., 1998; Young and Campbell, 1999; Scheuer, 2002; Young et al., 2006). cEEG recording is also performed in a commercially available computer-automated fashion to monitor depth of sedation and anaesthesia in humans (Bauerle et al., 2004; Kreuer et al., 2004). Some research in this latter area has been performed in beagles (Lee et al., 2012).
Acknowledgements
Illustrations prepared by A.D.K. James.
References
Accatino, A., Jaggy, A., Gaillard, C. and Aeschbacher, G. (1997) Electroencephalographic findings of encephalitis in beagle dogs experimentally infected with canine distemper virus (CDV). Journal of Veterinary Medicine, Series B 44, 39–48.
Akos, P., Thalhammer, J.G., Leschnik, M. and Halász, P. (2012) Electroencephalographic examination of epileptic dogs under propofol restraint. Acta Veterinaria Hungarica 60, 309–324.
Alarcon, G., Guy, C.N., Binnie, C.D., Walker, S.R., Elwes, R.D. and Polkey, C.E. (1994) Intracerebral propagation of interictal activity in partial epilepsy: implications for source localisation. Journal of Neurology, Neurosurgery, and Psychiatry 57, 435–449.
Amzica, F. (2002) Physiology of sleep and wakefulness as it relates to the physiology of epilepsy. Journal of Clinical Neurophysiology 19, 488–503.
Averill, D.R. and de Lahunta, A. (1968) Effects of some central nervous system depressants on the normal canine electroencephalogram. The Cornell Veterinarian 48, 620–632.
Badawy, R.A.B., Harvey, A.S. and Macdonell, R.A.L. (2009) Cortical hyperexcitability and epileptogenesis: Understanding the mechanisms of epilepsy - part 2. Journal of Clinical Neuroscience 16, 485–500. Ball, G., Gloor, P. and Schaul, N. (1977) The cortical electromicrophysiology of pathological delta waves in the electroencephalogram of cats. Electroencephalography and Clinical Neurophysiology 43, 346.
Bauerle, K., Greim, C.-A., Schroth, M., Geisselbrecht, M., Köbler, A. and Roewer, N. (2004) Prediction of depth of sedation and anaesthesia by the Narcotrend EEG monitor. British Journal of Anaesthesia 92, 841–845.
Berg, A.T., Berkovic, S.F., Brodie, M.J., Buchhalter, J., Cross, J.H., van Emde Boas, W., Engel, J., French, J., Glauser, T.A., Mathern, G.W., Moshé, S.L., Nordli, D., Plouin, P. and Scheffer, I.E. (2010) Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 51, 676–685.
Bergamasco, L., Accatino, A., Priano, L., Neiger-Aeschbacher, G., Cizinauskas, S. and Jaggy, A. (2003) Quantitative electroencephalographic findings in beagles anaesthetized with propofol. The Veterinary Journal 166, 58–66.
Electroencephalography
Blume, H.W. and Schomer, D.L. (1988) Surgical Approaches to Epilepsy. Annual Review of Medicine 39, 301–313.
Blume, W.T. (1997) Epilepsy: advances in management. European Neurology 38, 198–208.
Bouyer, J.J., Montaron, M.F. and Rougeul, A. (1981) Fast fronto-parietal rhythms during combined focused attentive behaviour and immobility in cat: cortical and thalamic localizations. Electroencephalography and Clinical Neurophysiology 51, 244–252.
Bouyer, J.J., Montaron, M.F., Vahnée, J.M., Albert, M.P. and Rougeul, A. (1987) Anatomical localization of cortical beta rhythms in cat. Neuroscience 22, 863–869.
Brauer, C., Kästner, S.B.R., Schenk, H.C., Tünsmeyer, J. and Tipold, A. (2011) Electroencephalographic recordings in dogs: Prevention of muscle artifacts and evaluation of two activation techniques in healthy individuals. Research in Veterinary Science 90, 306–311.
Brauer, C., Kästner, S.B.R., Rohn, K., Schenk, H.C., Tünsmeyer, J. and Tipold, A. (2012) Electroencephalographic recordings in dogs suffering from idiopathic and symptomatic epilepsy: diagnostic value of interictal short time EEG protocols supplemented by two activation techniques. The Veterinary Journal 193, 185–192.
Cascino, G. (2002) Video-EEG Monitoring in Adults. Epilepsia 43(Suppl. 3), 80–93.
Castellaro, C., Favaro, G., Castellaro, A., Casagrande, A., Castellaro, S., Puthenparampil, D.V. and Fattorello Salimbeni, C. (2002) An artificial intelligence approach to classify and analyse EEG traces. Clinical Neurophysiology 32, 193–214.
Caton, R. (1875) The Electric Currents of the Brain. The British Medical Journal 2, 278.
Chang, B.S. and Drislane, F.W. (2007) Epileptiform Abnormalities. In: Blum, A.S. and Rutkove, S.B. (eds) The Clinical Neurophysiology Primer. Humana Press Inc., Totowa, New Jersey, pp. 101–125.
Charles, M.S. and Fuller, J.L. (1956) Developmental study of the electroencephalogram of the dog. Electroencephalography and Clinical Neurophysiology 8, 645–652.
Chatrian, G.E., Bergamini, L., Dondey, M., Klass, D.W., Lennox-Buchthal, M. and Petersen, I. (1974) A glossary of terms most commonly used by clinical electroencephalographers. Electroencephalography and Clinical Neurophysiology 37, 538–548.
Croft, P.G. (1962) The EEG as an Aid to Diagnosis of Nervous Diseases in the Dog and Cat. The Journal of Small Animal Practice 3, 205–213.
Croft, P.G. (1970a) Electroencephalography in canine encephalitis. The Journal of Small Animal Practice 11, 241–249.
Croft, P.G. (1970b) Electroencephalography in canine head injury. The Journal of Small Animal Practice 11, 473–484.
Croft, P.G. (1971) Electroencephalography in cerebrovascular disease in small animals. The Journal of Small Animal Practice 12, 289–296.
Croft, P.G. (1972) Electroencephalography and space-occupying lesions in small animals. The Journal of Small Animal Practice 13, 175–184.
Cuffin, B.N., Schomer, D.L., Ives, J.R. and Blume, H. (2001a) Experimental tests of EEG source localization accuracy in realistically shaped head models. Clinical Neurophysiology 112, 2288–2292.
Cuffin, B.N., Schomer, D.L., Ives, J.R. and Blume, H. (2001b) Experimental tests of EEG source localization accuracy in spherical head models. Clinical Neurophysiology 112, 46–51.
Ebner, A., Sciarretta, G., Epstein, C.M. and Nuwer, M. (1999) EEG instrumentation. The International Federation of Clinical Neurophysiology. Electroencephalography and Clinical Neurophysiology Supplement 52, 7–10.
Engel, J., Burchfiel, J., Ebersole, J., Gates, J., Gotman, J., Homan, R., Ives, J., King, D., Lieb, J. and Sato, S. (1993) Long-term monitoring for epilepsy. Report of an IFCN committee. Electroencephalography and Clinical Neurophysiology 87, 437–458.
Farber, N.E., Poterack, K.A. and Schmeling, W.T. (1997) Dexmedetomidine and halothane produce similar alterations in electroencephalographic and electromyographic activity in cats. Brain Research 774, 131–141.
Fountain, N. (2006) EEG Is an Essential Clinical Tool: Pro and Con. Epilepsia 47(Suppl. 1), 23–25.
Fox, M.W. (1967a) Postnatal development of the EEG in the dog. I. Introduction and EEG techniques. The Journal of Small Animal Practice 8, 71–76.
Fox, M.W. (1967b) Postnatal development of the EEG in the dog. II. Development of electrocortical activity. The Journal of Small Animal Practice 8, 77–107.
Fox, M.W. (1967c) Postnatal development of the EEG in the dog. III. Summary and discussion of development of canine EEG. The Journal of Small Animal Practice 8, 109–112.
Hasegawa, D., Nakamura, S., Fujita, M., Takahashi, K. and Orima, H. (2005) A Dog Showing Klüver-Bucy Syndrome-like Behaviour and Bilateral Limbic Necrosis After Status Epilepticus. Veterinary Neurology and Neurosurgery Journal 7, 1–14.
Herin, R.A., Purinton, P.T. and Fletcher, T.F. (1968) Electroencephalography in the unanesthetized dog. American Journal of Veterinary Research 29, 329–336.
Hogan, K. and Fitzpatrick, J. (1988) The cerebral origin of the alpha rhythm. Electroencephalography and Clinical Neurophysiology 69, 79–81.
Holliday, T.A. and Williams, D.C. (1998) Interictal paroxysmal discharges in the electroencephalograms of epileptic dogs. Clinical Techniques in Small Animal Practice 13, 132–143.
Holliday, T.A. and Williams, D.C. (1999) Clinical Electroencephalography in Dogs. Veterinary Neurology and Neurosurgery Journal, 1(1), 1–38.
Holliday, T.A. and Williams, D.C. (2001) Advantages of Digital Electroencephalography in Clinical Veterinary Medicine Part 1. Veterinary Neurology and Neurosurgery Journal 3, 1–11.
Holliday, T.A. and Williams, D.C. (2003) Advantages of Digital Electroencephalography in Clinical Veterinary Medicine-2. Veterinary Neurology and Neurosurgery Journal 5, 1–16.
Holliday, T.A., Cunningham, J.G. and Gutnick, M.J. (1970) Comparative clinical and electroencephalographic studies of canine epilepsy. Epilepsia 11, 281–292.
Hori, M., Ito, T., Yoshida, K. and Shimizu, M. (1979) Effect of anticonvulsants on spiking activity induced by cortical freezing in cats. Epilepsia 20, 25–36.
Ives, J.R. (2005) New chronic EEG electrode for critical/intensive care unit monitoring. Journal of Clinical Neurophysiology 22, 119–123.
Ives, J.R., Rotenberg, A., Poma, R., Thut, G. and Pascual-Leone, A. (2006) Electroencephalographic recording during transcranial magnetic stimulation in humans and animals. Clinical Neurophysiology 117, 1870–1875.
Jaggy, A. and Bernardini, M. (1998) Idiopathic epilepsy in 125 dogs: a long-term study. Clinical and electroencephalographic findings. The Journal of Small Animal Practice 39, 23–29.
James, F.M.K., Allen, D.G., Bersenas, A.M.E., Grovum, W.L., Kerr, C.L., Monteith, G., Parent, J.M. and Poma, R. (2011) Investigation of the use of three electroencephalographic electrodes for long-term electroencephalographic recording in awake and sedated dogs. American Journal of Veterinary Research 72, 384–390.
Jasper, H.H. (1957) The Ten Twenty Electrode System of the International Federation. Electroencephalography and Clinical Neurophysiology 10, 371–375.
Kamp, A., Pfurtscheller, G. and Edlinger, G. (2005) Technological Basis of EEG Recording. In: Schomer, D.L. and Lopes da Silva, F. (eds) Niedermeyer’s Electroencephalography: Basic Principles, Clinical Applications, and Related Fields, 5th edn. Lippincott Williams & Wilkins, Philadelphia, Pennsylvania, pp. 127–138.
Kaplan, P.W. (2004) The EEG in metabolic encephalopathy and coma. Journal of Clinical Neurophysiology 21, 307–318.
Kaplan, P.W. (2006) The EEG of status epilepticus. Journal of Clinical Neurophysiology 23, 221–229.
Kellaway, P. (2003) Orderly approach to visual analysis: elements of the normal EEG and their characteristics in children and adults. In: Ebersole, J.S. and Pedley, T.A. (eds) Current Practice of Clinical Electroencephalography, 3rd edn. Lippincott Williams & Wilkins, Philadelphia, Pennsylvania, pp. 101–159.
King, A.S. (1987) Physiological and Clinical Anatomy of the Domestic Mammals: Central Nervous System (Vol. 1), 1st edn. Oxford University Press, Oxford, UK.
Klem, G. (2003) Artifacts. In: Ebersole, J.S. and Pedley, T.A. (eds) Current Practice of Clinical Electroencephalography, 3rd edn. Lippincott Williams & Wilkins, Philadelphia, Pennsylvania, pp. 271–281.
Klemm, W.R. (1965) Technical aspects of electroencephalography in animal research. American Journal of Veterinary Research 26, 1237–1248.
Klemm, W.R. (1968) Attempts to standardize veterinary electroencephalographic techniques. American Journal of Veterinary Research 29, 1895–1900.
Klemm, W.R. and Mallo, G.L. (1966) Clinical electroencephalography in anesthetized small animals. Journal of the American Veterinary Medical Association 148, 1038–1042.
Kreuer, S., Wilhelm, W., Grundmann, U., Larsen, R. and Bruhn, J. (2004) Narcotrend index versus bispectral index as electroencephalogram measures of anesthetic drug effect during propofol anesthesia. Anesthesia and Analgesia 98, 692–697.
Kusters, A.H., Vijn, P.C., van den Brom, W.E., Haberham, Z.L., Venker-van Haagen, A.J. and Hellebrekers, L.J. (1998) EEG-burst-suppression-controlled propofol anesthesia in the dog. The Veterinary Quarterly, 20(Suppl. 1), S105–106.
Lee, S.-H., Park, H.-W., Kim, M.-J., Noh, M.-H., Yoon, H.-S., Choi, B.-M., Lee, E.-K. and Noh, G.-J. (2012) External validation of pharmacokinetic and pharmacodynamic models of microemulsion and long-chain
Electroencephalography
triglyceride emulsion propofol in beagle dogs. Journal of Veterinary Pharmacology and Therapeutics 35, 329–341.
Lesser, R.P. and Webber, W.R.S. (2008) Seizure detection: reaching through the looking glass. Clinical Neurophysiology 119, 2667–2668.
Lewis, M.J., Williams, D.C. and Vite, C.H. (2011) Evaluation of the electroencephalogram in young cats. American Journal of Veterinary Research 72, 391–397.
Litt, B. and Cranstoun, S. (2003) Engineering principles. In: Ebersole, J.S. and Pedley, T.A. (eds) Current Practice of Clinical Electroencephalography, 3rd edn. Lippincott Williams & Wilkins, Philadelphia, Pennsylvania, pp. 32–71.
Litt, B. and Echauz, J. (2002) Prediction of epileptic seizures. Lancet Neurology 1, 22–30.
McConnell, J., Kirby, R. and Rudloff, E. (2007) Administration of acepromazine maleate to 31 dogs with a history of seizures. Journal of Veterinary Emergency and Critical Care 17, 262–267.
Mirsattari, S.M., Ives, J.R., Bihari, F., Leung, L.S., Menon, R.S. and Bartha, R. (2005) Real-time display of artifact-free electroencephalography during functional magnetic resonance imaging and magnetic resonance spectroscopy in an animal model of epilepsy. Magnetic Resonance in Medicine 53, 456–464.
Mormann, F., Andrzejak, R.G., Elger, C.E. and Lehnertz, K. (2007) Seizure prediction: the long and winding road. Brain 130, 314–333.
Niedermeyer, E. (2005) Maturation of the EEG: development of waking and sleep patterns. In: Schomer, D.L. and Lopes da Silva, F. (eds) Niedermeyer’s Electroencephalography: Basic Principles, Clinical Applications, and Related Fields, 5th edn. Lippincott Williams & Wilkins, Philadelphia, Pennsylvania, pp. 209–234.
Niedermeyer, E., Sherman, D.L., Geocadin, R.J., Hansen, H.C. and Hanley, D.F. (1999) The burst-suppression electroencephalogram. Clinical Electroencephalography 30, 99–105.
Noachtar, S., Binnie, C., Ebersole, J.S., Mauguiere, F., Sakamoto, A. and Westmoreland, B. (1999) A glossary of terms most commonly used by clinical electroencephalographers and proposal for the report form for the EEG findings. The International Federation of Clinical Neurophysiology. Electroencephalography and Clinical Neurophysiology Supplement 52, 21–41.
Nordli, D., Jr (2006) Usefulness of Video-EEG Monitoring. Epilepsia 47(Suppl. 1), 26–30.
Nuwer, M.R., Daube, J., Fischer, C., Schramm, J. and Yingling, C.D. (1993) Neuromonitoring during surgery. Report of an IFCN Committee. Electroencephalography and Clinical Neurophysiology 87, 263–276.
Nuwer, M.R., Comi, G., Emerson, R., Fuglsang-Frederiksen, A., Guérit, J.M., Hinrichs, H., Ikeda, A., Luccas, F.J. and Rappelsburger, P. (1998) IFCN standards for digital recording of clinical EEG. International Federation of Clinical Neurophysiology. Electroencephalography and Clinical Neurophysiology 106, 259–261.
Pampiglione, G. (1963) Development of Cerebral Function in the Dog. Butterworths, London.
Pellegrino, F. and Sica, R. (2004) Canine electroencephalographic recording technique: findings in normal and epileptic dogs. Clinical Neurophysiology 115, 477–487.
Poma, R., Ochi, A. and Cortez, M.A. (2010) Absence seizures with myoclonic features in a juvenile Chihuahua dog. Epileptic Disorders 12, 138–141.
Raith, K., Steinberg, T. and Fischer, A. (2010) Continuous electroencephalographic monitoring of status epilepticus in dogs and cats: 10 patients (2004-2005). Journal of Veterinary Emergency and Critical Care 20, 446–455.
Redding, R. and Knecht, C. (1984) Atlas of Electroencephalography in the Dog and Cat. Praeger Publishers, New York.
Redman, H.C., Wilson, G.L. and Hogan, J.E. (1973) Effect of chlorpromazine combined with intermittent light stimulation on the electroencephalogram and clinical response of the Beagle dog. American Journal of Veterinary Research 34, 929–936.
Reilly, E. (2005) EEG Recording and Operation of the Apparatus. In: Schomer, D.L. and Lopes da Silva, F. (eds) Niedermeyer’s Electroencephalography: Basic Principles, Clinical Applications, and Related Fields, 5th edn. Lippincott Williams & Wilkins, Philadelphia, Pennsylvania, pp. 139–160.
Roth, S.R., Sterman, M.B. and Clemente, C.D. (1967) Comparison of EEG correlates of reinforcement, internal inhibition and sleep. Electroencephalography and Clinical Neurophysiology 23, 509–520.
Scheuer, M.L. (2002) Continuous EEG monitoring in the intensive care unit. Epilepsia 43(Suppl. 3), 114–127.
Schubert, T.A., Chidester, R.M. and Chrisman, C.L. (2011) Clinical characteristics, management and long-term outcome of suspected rapid eye movement sleep behaviour disorder in 14 dogs. The Journal of Small Animal Practice 52, 93–100.
So, E.L. (2006) Value and limitations of seizure semiology in localizing seizure onset. Journal of Clinical Neurophysiology 23, 353–357.
Speckmann, E. and Elger, C. (2005) Introduction to the Neurophysiological Basis of the EEG and DC Potentials. In: Schomer, D.L. and Lopes da Silva, F. (eds) Niedermeyer’s Electroencephalography: Basic Principles, Clinical Applications, and Related Fields, 5th edn. Lippincott Williams & Wilkins, Philadelphia, Pennsylvania, pp. 17–29.
Steiss, J.E. (1988) A survey of current techniques in veterinary electrodiagnostics: EEG, spinal evoked and brainstem auditory evoked potential recording. Veterinary Research Communications 12, 281–288.
Steriade, M., Gloor, P., Llinás, R.R., Lopes da Silva, F. and Mesulam, M. (1990) Report of IFCN Committee on Basic Mechanisms. Basic mechanisms of cerebral rhythmic activities. Electroencephalography and Clinical Neurophysiology 76, 481–508.
Sundaram, M., Sadler, R.M., Young, G.B. and Pillay, N. (1999) EEG in epilepsy: current perspectives. The Canadian Journal of Neurological Sciences 26, 255–262.
Tobias, K.M., Marioni-Henry, K. and Wagner, R. (2006) A retrospective study on the use of acepromazine maleate in dogs with seizures. Journal of the American Animal Hospital Association 42, 283–289.
Tourai, K., Senba, H., Sasaki, N., Tokuriki, M., Ohashi, F., Takeuchi, A. and Usui, K. (1985) Developmental EEG of the beagle dog under xylazine sedation. Japanese Journal of Veterinary Science 47, 459–463.
Velis, D., Plouin, P., Gotman, J., Lopes da Silva, F. and ILAE DMC Subcommittee on Neurophysiology (2007) Recommendations Regarding the Requirements and Applications for Long-term Recordings in Epilepsy. Epilepsia 48, 379–384.
Wada, Y., Hasegawa, H., Okuda, H. and Yamaguchi, N. (1990) Anticonvulsant effects of zonisamide and phenytoin on seizure activity of the feline visual cortex. Brain and Development 12, 206–210.
Williams, D.C., Aleman, M., Holliday, T.A., Fletcher, D.J., Tharp, B., Kass, P.H., Steffey, E.P. and LeCouteur, R.A. (2008) Qualitative and quantitative characteristics of the electroencephalogram in normal horses during spontaneous drowsiness and sleep. Journal of Veterinary Internal Medicine 22, 630–638.
Wrzosek, M., Nicpon, J., Bergamasco, L., Sammartano, F., Cizinauskas, S. and Jaggy, A. (2009) Visual and quantitative electroencephalographic analysis of healthy young and adult cats under medetomidine sedation. The Veterinary Journal 180, 221–230.
Young, G.B. and Campbell, V.C. (1999) EEG monitoring in the intensive care unit: pitfalls and caveats. Journal of Clinical Neurophysiology 16, 40–45.
Young, G.B., Ives, J.R., Chapman, M.G. and Mirsattari, S.M. (2006) A comparison of subdermal wire electrodes with collodion-applied disk electrodes in long-term EEG recordings in ICU. Clinical Neurophysiology 117, 1376–1379.
12 Principles of Anti-epileptic Treatment
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Anti-epileptic treatment can be subdivided into:
Before initiating long-term anti-epileptic therapy, the clinician needs to ascertain that the animal has seizures rather than any other disorder that could mimic seizures (see Chapter 9), investigate (see Chapter 10), identify and treat (whenever possible) the underlying seizure aetiology. Sometimes a therapeutic trial will be necessary when the nature of the paroxysms is not known (e.g. seizure versus movement disorder), to see if the response supports the suspicion of seizure activity.
This chapter will focus on principles of long-term anti-epileptic therapy predominantly concerning pharmacologic treatment of idiopathic epilepsy. Treatment modalities including neurostimulation, dietary therapy, acupuncture, resective surgery and herbal medicine are presented in Chapter 25.
Specific treatments for various aetiologies of reactive seizure and structural epilepsy have been presented in Chapters 4 and 5 respectively.
Clinico-pathological investigations including haematology, serum biochemistry and urinalysis should be performed before initiation of anti-epileptic therapy to investigate for possible concurrent disorders that could alter anti-epileptic medication (AEM) disposition and to obtain a baseline for future monitoring of possible AEM-induced changes.
Aims of Anti-epileptic Treatment
The ideal goal of anti-epileptic therapy is to eradicate seizure activity. When this is not possible, we should aim to decrease seizure frequency, severity, duration and total seizure number (for animals that seizure in clusters) with no or acceptable AEM adverse effects in order to maximize the pet’s quality of life. To achieve these goals as much as is possible, the clinician needs to do the following.
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
When to Start Anti-epileptic Treatment
There are no evidence-based guidelines on when anti-epileptic treatment should be initiated in veterinary patients. Therefore this decision should be made on an individual basis depending on seizure type, frequency and aetiology, the general health of the patient, adverse effects associated with treatment, the clinician’s experience, and the pet owner’s life style and personal preference.
In general, in analogy with the guidelines developed in humans, it has been recommended to initiate long-term anti-epileptic treatment when the animal has:
In humans, initiation of anti-epileptic treatment after a first seizure decreases the risk of early seizure recurrence, but does not affect the long-term seizure remission rate. The risk of future seizures after two unprovoked seizures separated by at least 2 days is approximately 73% and therefore there is general consensus on starting anti-epileptic treatment after the second seizure (Musicco et al., 1997; Shih and ochoa, 2009; Glauser, 2013).
Some veterinary studies suggest that early treatment of idiopathic epilepsy results in a better long-term seizure control than delayed treatment (Heynold et al., 1997; Pakozdy et al., 2012). However, there is no consensus on optimal timing of treatment initiation based on number of isolated generalized or focal seizures that occur in a specific time frame. It has also been suggested to start anti-epileptic treatment when seizure frequency is unacceptable to the pet owner, however, this can be highly variable. In a questionnaire-based study on investigation of epileptic-dog owners’ perspective of seizure management the minimal seizure interval suggested as ‘reasonable’ varied from one seizure every 2 weeks to seizure free, with the most frequent response as one seizure every 3 to 6 months (Chang et al., 2006). In a larger questionnaire-based study on evaluation of quality of life in idiopathic epileptic dogs, the highest number of participants felt that only a seizure-free status is acceptable (38/128) and slightly fewer felt that one seizure every 3–6 months (27/128) or one seizure every 6 months (21/128) is acceptable (Wessmann et al., 2011).
In animals with metabolic or toxic disorders, anti-epileptic treatment (other than the short-term emergency treatment) may not be necessary when the underlying condition can be treated. However, with certain disorders short- to medium-term anti-epileptic treatment may be needed until the underlying disorder is permanently corrected (e.g. levetiracetam administration pre- and postoperatively in dogs with portosystemic shunts).
Choice of Anti-epileptic Medication
There are no evidence-based guidelines regarding the choice of AEM in dogs and cats. Therefore the AEM selection depends on several variables (Box 12.1). General guidelines on AEM selection are presented in Figs 12.1 and 12.2, however, AEM choice has to be done on an individual basis.
Since bromide (Br) was introduced by Sir Charles Locock in 1857, several AEMs have been developed; these have been categorized into first-, second-, third- and next-generation AEMs (Table 12.1) (Johannessen Landmark and Patselos, 2010; Podell, 2013). The slight discrepancies in the literature on the classification of second-, third-and next-generation AEMs are mainly due to differences in year of AEM approval in various countries. For simplicity, we will refer to them as new-generation AEMs in the rest of this chapter as opposed to first or old generation AEMs. The AEMs in bold in Table 12.1 are presented in Chapters 13 to 22.
First (or old) generation AEMs such as primidone, phenytoin, carbamazepine, valproate and ethosuximide are not discussed in


Box 12.1. Factors affecting anti-epileptic medication choice.
Medication-specific factors
individual chapters in this book due to their limited clinical use, inappropriate pharmacokinetics or toxicity in dogs and cats. Generally, in humans, new generation AEMs are safer, better tolerated, and have wider therapeutic ranges with reduced potential for drug interactions than first generation AEMs. Data on safety and efficacy of AEMs cannot be extrapolated from humans to dogs and cats as AEM metabolism, pharmacokinetics and pharmacodynamics may differ significantly among species. Information on new generation AEM safety, efficacy and pharmacokinetic interactions is limited in dogs and minimal to absent in cats. Some of the new AEMs such as vigabatrin, lamotrigine, tiagabine and oxcarbazepine have inappropriate pharmacokinetics or life-threatening adverse effects in dogs.
Mechanism of action, metabolism and pharmacokinetic interactions, pharmacokinetic parameters, dosage, time to steady state, reference serum concentration, adverse effects and efficacy of AEMs included in this book are summarized in Tables 12.2 to 12.10 and detailed in the respective chapters.
The ideal AEM should have high bioavailability, low and non-saturable protein binding, linear pharmacokinetics with clearance unaffected by renal impairment; additionally it should demonstrate rapid brain penetration, elimination half-life allowing once or twice daily dosing, no inhibition or induction of enzymatic biotransformation, low potential for pharmacokinetic interactions and adverse effects and high anti-epileptic efficacy (Stephen and Brodie, 2009; Podell, 2013). However, no such AEM currently exists. In addition, the ideal AEM should have antiepiletogenic activity (e.g. prevent, interrupt or reverse the epileptogenic process) or disease modifying properties (Kaminski et al., 2014).
Mechanisms of action of AEM involve modulation of ion channels, enhancement of inhibitory neurotransmission and attenuation of excitatory neurotransmission (Stephen and Brodie, 2009) (Table 12.2).
Table 12.1. Anti-epileptic medications categorized by generation of development.
Old generation New generation
| First generation | Second generation | Third generation | Next generation |
| (1957–1988) | (1989–2007) | (2008–2009) | (2010–2013) |
| Bromide | Felbamate | Lacosamide | Brivaracetam |
| Phenobarbital | Gabapentin | Rufinamide | Imepitoin (ELB138) |
| Benzodiazepines | Zonisamide | Eslicarbazepine | Carisbamate |
| Primidone | Levetiracetam | acetate | Flurofelbamate |
| Phenytoin | Pregabalin | Tiagabine | |
| Carbamazepine | Topiramate | Losigamone Retigabine | |
| Valproate | Lamotrigine | Remacemide | |
| Ethosuximide | Oxcarbazepine | Seletracetam | |
| Vigabatrin |
Table 12.2. Mechanisms of action of AEMs and related chapter in this book.
| Decreased | Decreased | Increased | Decreased | |||
|---|---|---|---|---|---|---|
| Na+ channel | Ca+ channel | GABA | glutamate | Novel | ||
| AEM | conductance | conductance | transmission | transmission | mechanism | Chapter |
| PB | ? | ++ | ? | 13 | ||
| Br | ++ | 14 | ||||
| ZNS | + | + (T-type) | ? | ? | 15 | |
| LEV | ? (N-type) | ? | ? | SV2A binding | 16 | |
| GBP | ++ (alpha2-delta) | ? | ? | 17 | ||
| PGB | ++ (alpha2-delta) | 17 | ||||
| FBM | + | + | ? | + | 18 | |
| TPR | + | + | + | + | 19 | |
| LCM | ++ | CRMP-2 binding | 20 | |||
| BRV | + | ? | SV2A binding | 20 | ||
| RFM | ++ | 20 | ||||
| BNZ | ++ | 21 | ||||
| IMP | + | ++ | 22 |
PB, phenobarbital; Br, bromide; ZNS, zonisamide; LEV, levetiracetam; GBP, gabapentin; PGB, pregabalin; FBM, felbamate; TPR, topiramate; LCM, lacosamide; BRV, brivaracetam; RFM, rufinamide; BNZ, benzodiazepine; IMP, imepitoin ++ primary mechanism of action; + probable action; ? possible action; SV2A, synaptic vesicle protein 2A; CRMP-2, collapsing response mediator protein-2
AEM Metabolism (% metabolized) Primary excretion Effect on pharmacokinetics of AEMs Comment
| PB | Hepatic microsomal cytochrome | Renal | Potent inducer of hepatic microsomal | Chronic PB administration can affect the | ||||
|---|---|---|---|---|---|---|---|---|
| P450 enzymes (75%) | cytochrome P450 enzymes leading to | disposition of other co-administered | ||||||
| ↑ Cl of hepatically metabolized | medications which are metabolized by | |||||||
| medications; | CPY450 and/or bound to plasma | |||||||
| ↑ Cl and ↓ T1/2 of PB itself; | α1-acid glycoprotein. | |||||||
| ↑ Cl and ↓ Cmax, ↓T1/2, of ZNS; | Concurrent administration of medications that | |||||||
| ↑ Cl and ↓ Cmax, ↓T1/2, of LEV; | inhibit hepatic microsomal CYP450 enzymes | |||||||
| ↑ Cl and ↓ Cmax of diazepam and oxazepam | may inhibit PB metabolism, increase serum | |||||||
| ↑ Cl and ↓ Cmax of clorazepate | concentration and result in toxicity. | |||||||
| (nordiazepam) | Ammonium chloride for urine alkalinization | |||||||
| ↑ Cl of voriconazole | increases renal elimination of PB. | |||||||
| Br | Not metabolized | Renal | None reported | The rate of elimination of Br varies | ||||
| proportionally and inversely to chloride | ||||||||
| intake. | ||||||||
| Loop diuretics (such as furosemide) may | ||||||||
| enhance Br elimination, whereas | ||||||||
| osmotic diuretics do not seem to affect | ||||||||
| Br excretion. | ||||||||
| ZNS | Hepatic (90%) | Renal | None reported | Caution is warranted when administered | ||||
| concurrently with other carbonic | ||||||||
| anhydrase inhibitors. | ||||||||
| LEV | Enzymatic hydrolysis by hydrolases, | Renal | No effect on steady-state serum | Concurrent PB administration ↑ Cl and | ||||
| amidases and β-esterases in the | concentration of PB and Br | ↓ Cmax, ↓T1/2, of LEV | ||||||
| bloodstream, liver and other tissues, | ||||||||
| and oxidation (38–50%) | ||||||||
| GBP | Hepatically metabolized to N-methyl- | Renal | Unknown in dogs. | In people: co-administration with antiacids | ||||
| gabapentin (30–40%) | In people: ↓ FBM Cl by 37% | containing aluminium or magnesium can | ||||||
| and ↑FBM T1/2 by 46% | ↓ GBP absorption by up to 24%. | |||||||
| Co-administration with cimetidine can | ||||||||
| ↓ GBP renal clearance by 14% | ||||||||
| PGB | Unknown in dogs. | Renal (in people). | Unknown in dogs. | In humans, PGB concentrations ↓ by 20% | ||||
| In humans, no metabolism, 98% | Unknown in dogs | In humans, PGB co-administration did not | to 30% following co-administration with | |||||
| excreted unchanged in the urine | alter the steady-state plasma concentrations | enzyme-inducing AEMs | ||||||
| of PB, TPR and other AEMs | ||||||||
| Continued | ||||||||
Principles of Anti-epileptic Treatment
| AEM | Metabolism (% metabolized) | Primary excretion | Effect on pharmacokinetics of AEMs | Comment |
| FBM | Hepatic microsomal cytochrome P450 enzymes | Renal | Unknown in dogs. In people, ↑ serum PB concentration | In people, FBM Cl ↑ with concurrent administration of PB and ↓ by 37% |
| by 24% | with concurrent administration of GBP | |||
| TPR | Unknown in dogs. | Renal (in humans). | Unknown in dogs. | Multiple TPR dosing produced no |
| In humans, hepatically | Unknown in dogs | Relatively low potential for clinically relevant | autoinduction or inhibition of enzymes | |
| metabolized (50%) | interactions with other medications. Does | that metabolize TPR | ||
| not seem to significantly alter PB | ||||
| pharmacokinetics in humans | ||||
| LCM | Unknown in dogs. In humans, metabolized by | Renal (in people). Unknown in dogs | Unknown in dogs. In humans, no relevant influence on plasma | In humans, LCM serum concentration ↓ by 15–20% with concurrent |
| CYP2C19 (60%) | concentrations of PB, LEV, GBP, TPR and | administration of enzyme-inducing | ||
| other AEMs | AEMs | |||
| BRV | Unknown in dogs. | Renal (in humans). | Unknown in dogs | Unknown |
| In humans, hydrolysis and secondarily | Unknown in dogs | |||
| hydroxylation mediated by CYP 2C19 | ||||
| RFN | Unknown in dogs. In humans, extensively metabolized by | Renal (in humans). Unknown in dogs | Unknown in dogs. In humans, RFN does not affect | In people, concurrent administration of PB, ↓ plasma RFN concentration |
| carboxyesterase-mediated enzymatic | trough concentrations of PB, TPR | by up to 46.3% | ||
| hydrolysis in the liver | and other AEMs | |||
| IMP | Hepatic oxidation to four | Faecal | Unknown | No harmful clinical interactions were |
| major inactive metabolites | observed when IMP was used as | |||
| adjunctive treatment to PB or PRM in a | ||||
| small number of epileptic dogs. |
352 L. De Risio
PB, phenobarbital; Br, bromide; ZNS, zonisamide; LEV, levetiracetam; GBP, gabapentin; PGB, pregabalin; FBM, felbamate; TPR, topiramate; LCM, lacosamide; BRV, brivaracetam; RFM, rufinamide; IMP, imepitoin; PRM, primidone; CYP, cytochrome; Cl, clearance; T1/2, elimination half-life; C, maximum concentration; ↑, increases; ↓, decreases
max
| Serum or | ||||||||
|---|---|---|---|---|---|---|---|---|
| Dose administered in | plasma protein | |||||||
| AEM | pharmacokinetic study | Oral F (%) | Vd (l/kg) | binding (%) | Cmax (mg/l)a | Tmax (h) | T1/2 (h)b | Reference |
| PB | 10 mg/kg PO once | 91 (86–96) | 0.7 ± 0.15* | 45–46 | 15 (14–17) | 6 (4–8) | 52 (44–62) | Al-Tahan, 1985; | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| *Pedersoli, 1987 | |||||||||||||||||
| 5.5 mg/kg PO once | NA | NA | NA | 8.94 ± 1.67 | 3.80 ± 1.48 | 88.7 ± 19.6 | Ravis, 1989 | ||||||||||
| 5.5 mg/kg/day PO | NA | NA | NA | 27.69 ± 4.04 | 4.60 ± 0.89 | 47.3 ± 10.7 | |||||||||||
| for 90 d | |||||||||||||||||
| Br | 20 mg/kg PO once | 46 | 0.45 ± 0.07 | 0 | 79 ± 13 | 0.69 ± 0.13 | 46 ± 9 days Trepanier, 1995b | ||||||||||
| ZNS | 10.3 mg/kg PO once | 68 ± 12 | 0.73 ± 0.15 | 40 ± 13 | 13.2 ± 2.0 | 2.5 ± 0.65 | 17.4 ± 4.9 | Boothe, 2008 | |||||||||
| 10.3 mg/kg q12h PO | NA | NA | NA | 52 ± 8.7 | NA | 21.4 ± 5.4 | Boothe, 2008 | ||||||||||
| for 8 weeks | |||||||||||||||||
| 5 mg/kg once | NA | NA | NA | 3.8 | 6 | 13 | Orito, 2008 | ||||||||||
| LEV | 54 mg/kg PO once | NA | NA | <10 | NA | NA | 2.3–3.1 | Benedetti, et al., 2004 | |||||||||
| 20 mg/kg PO once | NA | NA | NA | 59.91 ± 11.54 | 0.62 | 2.87 ± 0.21 | Moore, 2010 | ||||||||||
| 20 mg/kg PO q8h | NA | NA | NA | 52.41 ± 10.08 | 1 | 3.59 ± 0.82 | |||||||||||
| for 7 days | |||||||||||||||||
| 20 mg/kg PO once | 100 | 0.59 | NA | 30 ± 4 | 1.4 ± 0.5 | 3 ± 0.3 | Patterson, 2008 | ||||||||||
| 20 mg/kg IM once | 113 | 0.60 | NA | 30 ± 3 | 0.7 ± 0.3 | 3 ± 0.4 | |||||||||||
| 20 mg/kg IV once | NA | 0.55 | NA | 37 ± 5 | NA | 3 ± 0.3 | |||||||||||
| 60 mg/kg IV once | NA | 0.48 ± 0.08 | NA | 254 ± 81 | NA | 4.0 ± 0.82 | Dewey, 2008 | ||||||||||
| 40 mg/kg rectally | NA | NA | NA | 222 ± 72 | 36.0 ± 10.7 | NA | Peters, 2014 | ||||||||||
| GBP | 10 mg/kg PO once | NA | 0.942 (0.85–1.256) | NA | 9.68 (5.32–10.90) | 1.5 (0.75–2) | 3.35 (2.63–3.68) KuKanich, 2011 | ||||||||||
| 20 mg/kg PO once | NA | 1.207 (0.835–1.476) | NA | 12.95 (10.7–18.2) | 1.75 (1–2) | 3.39 (3.07–3.91) KuKanich, 2011 | |||||||||||
| 50 mg/kg PO once | 80 | NA | < 3 | 56.3 | 1.1 | 2.2 | Radulovic, 1995 | ||||||||||
| PGB | 2 mg/kg PO once | NA | NA | NA | 5 (4.1–5.9) | 0.75 (0.5–1) | 6.1 (5.79–6.38) | Salazar, 2009 | |||||||||
| 4 mg/kg PO once | NA | NA | NA | 7.15 (4.6–7.9) | 1.5 (1.0–4.0) | 6.90 (6.21–7.40) Salazar, 2009 | |||||||||||
| Continued | |||||||||||||||||
Principles of Anti-epileptic Treatment
Table 12.4. Continued.
| Serum or | ||||||||
|---|---|---|---|---|---|---|---|---|
| Dose administered in | plasma protein | |||||||
| AEM | pharmacokinetic study | Oral F (%) | Vd (l/kg) | binding (%) | Cmax (mg/l)a | Tmax (h) | T1/2 (h)b | Reference |
| FBM | 20 mg/kg PO once | NA | NA | NA | 48.6 ± 8.4 | 4.8 ± 2.3 | 6.7 ± 1.6 | Adusumalli, 1992 |
| 20 mg/kg PO q24h | NA | NA | NA | 45.9 ± 10.3 | 3.6 ± 1.3 | 5.24 ± 1.1 | ||
| for 10d | ||||||||
| TPR | 40mg/kg PO once | 27–59 | NA | 8–13 | 45.4 ± 10.8 | 1.4 ± 0.3 | 2.6 ± 0.3 | Streeter, 1995 |
| 40mg/kg PO q24h | NA | NA | NA | 43.5 ± 9.2 | 1.1 ± 0.3 | 2.0 ± 0.1 | ||
| for 15d | ||||||||
| 10 mg/kg IV once | NA | 0.63 | NA | NA | NA | NA | ||
| LCM | 3 mg/kg PO once | NA | 0.402 ± 0.122 | NA | 8.06 ± 5.46 | 2.33 ± 1.44 | 3.58 ± 1.40 | Martinez, 2012 |
| RFM | 20 mg/kg PO once | NA | 1.25 | NA | 19.6 ± 5.8 | 9.33 ± 4.68 | 9.86 ± 4.77 | Wright, 2012 |
| IMP | 30 mg/kg q12h | 92 | 1.2 ± 0.6 | 60–70 | 15.6 ± 5.2 | 1.8 ± 0.5 | 1.7 ± 0.6 | Boehringer Ingelheim |
| (0.4–2.5) | (7.6–28.3) | (1–3) | (0.8–2.9) | data on file 2008 |
aThe value indicated in mg/l is unchanged in µg/ml bExcept Br whose T1/2 is expressed in days PB, phenobarbital; Br, bromide; ZNS, zonisamide; LEV, levetiracetam; GBP, gabapentin; PGB, pregabalin; FBM, felbamate; TPR, topiramate; LCM, lacosamide; RFM, rufinamide; IMP, imepitoin; PO, per os; F, Bioavailability; Vd, Volume of distribution; h, hours; d, days; C, maximum concentration; T, time to maximum concentration; T1/2, elimination half-life;
maxmax
NA, not assessed
354 L. De Risio
Table 12.5. Pharmacokinetic parameters of AEMs following oral administration in healthy cats.
| Dosage administered | ||||||||
|---|---|---|---|---|---|---|---|---|
| in pharmacokinetic | Serum protein | |||||||
| AEM | study | Oral F (%) | Vd (l/kg) | binding (%) | Cmax (mg/l)a | Tmax | T1/2 | Reference |
| PB | 10 mg/kg PO once | 120 ± 0.12 | 0.73 ± 0.04 | NA | NA | NA | 76.1 ± 6.96hb | Cochrane, 1990a |
| 5 mg/kg PO once | NA | 0.77 ± 0.02 | NA | NA | NA | 47.6 ± 2.89hb | Cochrane, 1990b | |
| 5 mg/kg/d PO for 21 d | NA | 0.70 ± 0.04 | NA | NA | NA | 43.3 ± 2.92hb | ||
| KBr | 15 mg/kg PO q12h | NA | NA | 0 | 1100 ± 200 | 56 days | 12 days | Boothe et al., 2002 |
| for 81 weeks | ||||||||
| ZNS | 10 mg/kg once | NA | NA | NA | 13.1 (10.1–14.3) | 4 (2–8) | 33 (21.3–44.8) | Hasegawa et al., 2008 |
| 20 mg/kg/day for | NA | NA | NA | NA | 59 | NA | ||
| 9 weeks | ||||||||
| LEV | 20 mg/kg PO once | 102 ± 39 | NA | NA | 25.54 ± 7.97 | 1.67 ± 1.73 | 2.95 ± 0.95 | Carnes et al., 2011 |
| 20 mg/kg IV once | NA | 0.52 ± 0.09 | NA | 37.52 ± 6.79 | NA | 2.86 ± 0.65 | ||
| GBP | 10 mg/kg PO once | 88.7 ± 11.1 | 0.65 ± 0.014c | NA | 7.982 ± 1.053 | 1.67 ± 0.37 | 2.83 ± 0.34 | Siao et al., 2010 |
| (49.6–118.3) | (4.638–10.550) | (0.97–2.92) | (2.52–3.30) hc | |||||
| DZP | 10 mg/kg IV once | NA | 3.07 ± 1.6 | 94% | NA | NA | 5.5 (diazepam) | Cotler et al., 1984 |
| 21.3 (nordiazepam) |
aThe value indicated in mg/l is unchanged in µg/ml bExcept for Br after administration of 4mg/kg GBP IV cPB, phenobarbital; Br, bromide; ZNS, zonisamide; LEV, levetiracetam; GBP, gabapentin; DZP, diazepam; F, biovailability; Vd, volume of distribution; C, maximum concentration; T,
maxmax
time to maximum concentration; T½, elimination half-life
Principles of Anti-epileptic Treatment
Table 12.6. Recommended dosage, time to steady state, reference serum concentration, conversion factor, cost (at the time of writing) and related Chapter number of AEMs in dogs.
| Reference serum | Conversion | ||||
|---|---|---|---|---|---|
| concentration | factor (f) | ||||
| AEM | Dosage (PO) | Tss (days) | range mg/la | (µmol/l = f × mg/l) Cost | Chapter |
| PB | 2–3 mg/kg q12hb | 10–20 | 20–35 | 4.31 | $ | 13 |
| KBr | 30–40 mg/kg once or | 90–120c | 2000–3000 when | 0.0125 | $ | 14 |
| divided twice daily | Br is used as | |||||
| as monotherapy | monotherapy | |||||
| 20–30 mg/kg once or | 1000–2000 when | |||||
| divided twice daily | Br is used in | |||||
| as adjunctive therapy | combination with PB | |||||
| ZNS | 3–5 mg/kg q12h; | 3–4 | NE; 10–40d | 4.71 | $$ | 15 |
| 7–10 mg/kg q12h | ||||||
| (when administered | ||||||
| with PB) | ||||||
| LEV | 20 mg/kg q8h | 1 | NE; 5–45 or | 5.88 | $$ | 16 |
| 12–46d | ||||||
| GBP | 10–20 mg/kg q6–8h | 1 | NE; 2–20d | 5.84 | $ | 17 |
| PGB | 2–4 mg/kg q8–12h | 1–2 | NE; 2.8–8.3d | 6.28 | $$ | 17 |
| FBM | 20 mg/kg q8h | 1–2 | NE; 30–60d | 4.20 | $$$ | 18 |
| TPR | 2-10 mg/kg q12he | NE | NE; 5–20d | 2.95 | $$$ | 19 |
| IMP | 10–30 mg/kg q12h | 1–2 | NE | NE | $$ | 22 |
aThis value is the same in mg/l or µg/ml bThis is the initial recommended dosage. Dose increase is often required over time to maintain reference serum PB concentration. If T1/2 becomes shorter than 20 h administration q 8 h is required cAffected by chloride intake dProposed reference range in people eProposed in Kiviranta et al, 2013 (titration from 2 mg/kg q12h for 3 weeks to 5 mg/kg q12h and if inadequate seizure control further increase to 10 mg/kg q8-12h)
FBM, felbamate; GBP, gabapentin; h, hours; IMP, imepitoin; KBr, potassium bromide; LEV, levetiracetam; NE,not established in dogs; PB, phenobarbital; PGB, pregabalin; PO, per os; RFM, rufinamide; TPR, topiramate; Tss, time to steady state; ZNS, zonisamide; $, cheap, $$ moderate cost, $$$ very expensive
Table 12.7. Recommended dosage, reference serum concentration, conversion factor and related Chapter of AEMs in cats.
| Conversion factor (f) | |||||
|---|---|---|---|---|---|
| Reference serum | (µmol/l = f × mg/l) or | ||||
| AEM | Dosage (PO) | Tss (days) | concentration range | (µmol/l = f × ng/ml) | Chapter |
| PB | 1.5–2.5 mg/kg q12h | 10–15 | 15–30 mg/l | 4.31 | 13 |
| LEV | 20 mg/kg q8h | 1 | NE; 5–45 or | 5.87 | 16 |
| 12–46 mg/la | |||||
| DZP | 2 to 5 mg q8–12hb | 4 | 500–700 ng/ml | 0.0035 | 21 |
| (nordiazepam) | |||||
| GBP | 3–10 mg/kg q6–8h | 1 | NE; 2–20 mg/la | 5.84 | 17 |
| ZNS | 5–10 mg/kg q24hc | 7 | NE | 4.71 | 15 |
aProposed reference range in people bThis dosage can be increased by 2 mg at a time up to a maximum total daily dose of 20 mg cThis dosage has been proposed based on one pharmacologic study (Hasegawa et al., 2008) and has not been evaluated in the clinical setting DZP, diazepam; GBP, gabapentin; h, hours; LEV, levetiracetam; NE, not established in cats; PB, phenobarbital; PO, per os; Tss, time to steady state; ZNS, zonisamide
Table 12.8. Adverse effects of AEM in dogs and cats (uncommon adverse effects and possible idiosyncratic reactions are indicated in italic).
AEM Adverse effects in dogs Adverse effects in cats
| PB | Sedation | Sedation |
| Ataxia | Ataxia | |
| Polyphagia | Polyphagia | |
| Polydipsia/polyuria | Polydipsia/polyuria | |
| Hepatotoxicity | Thrombocytopenia | |
| Hematologic abnormalities (anaemia, | Marked sedation, vomiting, leucopenia, | |
| and/or thrombocytopenia | neutropenia, generalized pruritus and | |
| and/or neutropenia) | distal limb oedema | |
| Superficial necrolytic dermatitis | Facial pruritus | |
| Increased risk of pancreatitis | Generalized lymphadenopathy alone or | |
| Dyskinesia and anxiousness | with concurrent lethargy, anorexia, | |
| cutaneous and oral muco-cutanous | ||
| erythaema, erosions, ulcerations and | ||
| crusting | ||
| Br | Sedation | Lower airway disease |
| Ataxia and pelvic limb weakness | ||
| Polydipsia/polyuria | ||
| Polyphagia with weight gain | ||
| Nausea, vomiting and/or diarrhoea | ||
| Personality changes such as aggressive | ||
| behaviour, irritability and hyperactivity | ||
| Erythematous dermatitis | ||
| Panniculitis | ||
| Persistent cough | ||
| Increased risk of pancreatitis | ||
| ZNS | Sedation | Anorexia |
| Generalized ataxia | Further assessment of tolerability, safety | |
| Vomiting | and possible adverse effects of this | |
| Inappetence | AEM is required in this species | |
| Keratoconjunctivitis sicca | ||
| Polyarthropathy | ||
| Acute hepatopathy/hepatomegaly | ||
| Renal tubular acidosis | ||
| LEV | Sedation | Transient mild lethargy |
| Ataxia | Inappetence | |
| Decreased appetite or anorexia | ||
| Vomiting | ||
| Behavioural changes | ||
| GBP | Sedation | Further assessment of tolerability, safety |
| Ataxia | and possible adverse effects of this | |
| AEM is required in this species | ||
| PGB | Sedation | U |
| Ataxia | ||
| Dizziness | ||
| Weakness | ||
| FBM | Keratitis sicca | U |
| Thrombocytopenia | ||
| Lymphopenia and leucopenia | ||
| Further assessment of tolerability, safety | ||
| and possible adverse effects of this | ||
| AEM is required in this species | ||
| Continued |
| AEM | Adverse effects in dogs | Adverse effects in cats |
| TPR | No adverse effects were reported in healthy | U |
| beagle dogs administered 10–150 mg/kg | ||
| daily oral doses for 15 days. | ||
| Sedation, ataxia and weight loss as | ||
| adjunctive anti-epileptic medication in the clinical | ||
| setting | ||
| Further assessment of tolerability, safety | ||
| and possible adverse effects of this | ||
| AEM is required in this species | ||
| LCM | U | U |
| BRV | U | U |
| RFN | No short-term adverse effects were | U |
| reported in healthy dogs administered | ||
| a single oral dose of 20 mg/kg | ||
| Further assessment of tolerability, safety | ||
| and possible adverse effects of this | ||
| AEM is required in this species | ||
| IMP | Polyphagia | U |
| Hyperactivity, polyuria, polydipsia, somnolence, | ||
| hypersalivation, emesis, ataxia, lethargy, | ||
| diarrhoea, prolapsed nictitating membrane, | ||
| decreased vision and sensitivity to sound | ||
| DZP | Sedation | Hepatotoxicity (possibly idiosyncratic) |
| Paradoxical excitement/hyperactivity | ||
| Sedation |
PB, phenobarbital; Br, bromide; ZNS, zonisamide; LEV, levetiracetam; GBP, gabapentin; PGB, pregabalin; FBM, felbamate; TPR, topiramate; LCM, lacosamide; BRV, brivaracetam; RFM, rufinamide; IMP, imepitoin; DZP, diazepam; U, unknown
Initial anti-epileptic treatment
Initial anti-epileptic treatment generally involves the use of one AEM (monotherapy) as this is associated with lower cost, better compliance and less potential for adverse effects and pharmacokinetic or pharmacodynamic interactions (Stephen and Brodie, 2009; Muñana, 2013) (Figs 12.1 and 12.2). An exception to this general rule can be made when an AEM with a very long half-life such as Br is combined with a medication with short half-life such as levetiracetam; the latter is administered at least until Br steady-state reference serum concentrations are achieved. This initial combined treatment avoids the adverse effects associated with Br loading (see Chapter 14) while providing immediate anti-epileptic treatment. In addition, early dual-therapy (two AEMs) may be considered in idiopathic epileptic dogs with frequent and severe seizures (e.g. young Border collies, German shepherd dogs).
Phenobarbital (PB) is commonly the first AEM of choice in epileptic dogs and cats due to its pharmacokinetic profile, relative safety, affordable cost and efficacy (Fig. 12.1). The preferential use of PB, compared with Br, as first choice AEM in idiopathic epileptic dogs is supported by a recent double-blinded, randomized, parallel, clinical trial (Boothe et al., 2012; Table 12.9).
Imepitoin (IMP) (Chapter 22) has recently been introduced in Europe as treatment of generalized-onset seizures in idiopathic epileptic dogs. Its use is not recommended in dogs with hepatic dysfunction. Information on pharmacokinetic interactions is currently limited, however, the pharmacokinetic interaction potential seems low. In a recent multi-centre, randomized, blinded, parallel clinical trial in idiopathic epileptic dogs, there was no
| Number (percentage) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| of dogs with ≥50% | |||||||||||||||||
| Number | Number (percentage) | reduction in seizure | |||||||||||||||
| (percentage) of dogs that withdrew | frequency, with | Number | |||||||||||||||
| Number | of dogs that | from the study | respect to baseline | (percentage) Follow-up | |||||||||||||
| Type of | of dogs | completed | because of | (including | of seizure-free duration | ||||||||||||
| AEM | therapy | Study type | included | the study | adverse effects | seizure-free dogs) | dogs | (months) | Reference | ||||||||
| PB | M | Double-blinded, | 21 | 20 (95%) | 1 (5%) | 18 (90%) | 17 (85%) | 6 | Boothe et al., | |
|---|---|---|---|---|---|---|---|---|---|---|
| randomized, | 2012 | |||||||||
| parallela | ||||||||||
| Br | M | Double-blinded, | 25 | 23 (92%) | 2 (8%) | 15 (65%) | 12 (52%) | 6 | Boothe et al., | |
| randomized, | 2012 | |||||||||
| parallel | ||||||||||
| AT to PB | Retrospective | 22 | 19 (86%) | NA | 12 (63%) | 5 (26%) | 7–61 | Schwartz-Porsche | ||
| or PRM | open label | et al., 1990 | ||||||||
| AT to PB | Retrospective | 23 | 17 (74%) | NA | 11 (65%) | 3 (18%) | 12 | Podell and | ||
| open label | Fenner, 1993 | |||||||||
| M or AT to | Retrospective | 122 | NA | NA | 88 (72%) | NA | 4–60 | Trepanier et al., | ||
| PB or PRM | open label | (median 12) | 1998 | |||||||
| ZNS | M | Prospective | 10 | 10 (100%) | 0 | 6 (60%) | 3 (30%) | 2–36 | Chung et al., | |
| open label | 2012 | |||||||||
| AT to PB ± Br | Prospective | 12 | 12 (100%) | 0 | 7 (58%) | 2 (17%) | 2–18 | Dewey et al, | ||
| open label | 2004 | |||||||||
| AT to PB ± Br | Prospective | 10 AT | 11 (100%) | 0 | 8 (80%) AT | 2 (20%) AT | 4 | Von Klopman | ||
| or M | open label | 1 M | 1 (100%) M | et al, 2004 | ||||||
| LEV | AT to PB and Br Prospective | 14 | 14 (100%) | 0 | 8 (57%) | 3 (21%) | 2 | Volk et al, 2008 | ||
| open label | 14 | 11 (76%) | 1 (7%) | 7 (63%) | 1 (9%) | 6 | ||||
| AT to PB and Br Placebo | 34 | 28 (82%) | 2 | 16 (56%) | 5 (17%) | 4 | Munana et al, | |||
| controlled | 2012 | |||||||||
| randomized | ||||||||||
| blinded | ||||||||||
| Continued | ||||||||||
Principles of Anti-epileptic Treatment
| Number (percentage) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| of dogs with ≥50% | |||||||||||||||||
| Number | Number (percentage) | reduction in seizure | |||||||||||||||
| (percentage) of dogs that withdrew | frequency, with | Number | |||||||||||||||
| Number | of dogs that | from the study | respect to baseline | (percentage) Follow-up | |||||||||||||
| Type of | of dogs | completed | because of | (including | of seizure-free duration | ||||||||||||
| AEM | therapy | Study type | included | the study | adverse effects | seizure-free dogs) | dogs | (months) | Reference | ||||||||
| GBP | AT to PB ± Br | Prospective | 17 | 15 (88%) | 0 | 7 (46%) | 3 (20%) | 4 | Govendir et al., | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| open label | 2005 | |||||||||||
| AT to PB and Br Open label | 11 | 11 (100%) | 0 | 6 (55%) | 2 (18%) | 3 | Platt et al., 2006 | |||||
| PGB | AT to PB ± Br | Open label | 11 | 9 (82%) | 2 (18%) | 7 (78%) | 0 | 3 | Dewey et al., 2009 | |||
| FBM | M or AT to PB | Open labelb | 5 M | 6 (100%) | NA | 6 (100%) | 2 (33%) | 2–22 | Ruehlmann | |||
| 1 AT | et al., 2001 | |||||||||||
| IMP | M | Prospective | 12 | U | 0 | 4/12 (33%)c | 1/12 (8%) | 7.7 ± 0.7 | Rieck et al., 2006 | |||
| open label | ||||||||||||
| AT to PB | Prospective | 17 | U | 0 | 6/17 (35%)c | 1/17 (6%) | 5.6 ± 0.7 | Rieck et al., 2006 | ||||
| or PRM | open label | |||||||||||
| M | Prospective | 116 | 64 (55%) | NA | 48/64 (75%) | 30/64 (47%) | 4 | SPC, 2013 | ||||
| multicentre, | ||||||||||||
| randomized, | ||||||||||||
| blinded | ||||||||||||
| TPR | AT to PB ± Br | Prospective | 10 | 7 (70%) | 1 | 5/10 (50%) | 2/10 (20%) | 6 | Kiviranta et al., | |||
| open label | 2013 | |||||||||||
aA parallel designed clinical trial compares the results of two different treatments on two separate groups of patients bThis study included dogs with focal seizures only cNumber (percentage) of dogs with >50% reduction in seizure frequency during treatment PB, phenobarbital; Br, bromide; ZNS, zonisamide; LEV, levetiracetam; GBP, gabapentin; PGB, pregabalin; FBM, felbamate; IMP, imepitoin; PRM, primidone; TPR, topiramate; NA, not available; U, unknown
360 L. De Risio
| Number | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| (percentage) of | |||||||||
| cats with ≥50% | |||||||||
| Number | reduction in | ||||||||
| (percentage) | seizure | ||||||||
| Number | of cats that | frequency, | |||||||
| (percentage) | withdrew from | with respect | Number | ||||||
| of cats that | the study | to baseline | (percentage) | ||||||
| Type of | Number of | completed | because of | (including | of seizure-free | Follow-up | |||
| AEM | therapy | Study type | cats included | the study | adverse effects | seizure-free cats) | cats | duration | Reference |
| PB | M | Retrospective | 16 | NA | NA | (75%) | (44%) | NA | Volk et al., |
| open label | 2007 | ||||||||
| M or AT with LEV, | Open label | 36 | 28 (78%) | NA | (74%)a | (43%)a | 1–5 years | Pakozdy et al., | |
| DZP, GBP | 2012 | ||||||||
| M | Open label | 30 | 30 | 0 | 30 (100%) | 13 (43%) | 0.63-91 months | Finnerty, 2014 | |
| (median | |||||||||
| 14 months) | |||||||||
| LEV | AT to PB | Open label | 12 | 10 (83%) | 0 | 7 (70%) | 3 (30%) | 3 months | Bailey et al., |
| 2008 | |||||||||
| Brb | M or AT to PB | Retrospective | 15 | NA | 2 | NA | 7/15 (47%)c | NA | Boothe et al., |
| open label | 2002 |
aExpressed as mean over the entire study period bThe use of Br in cats is not recommended due to adverse effects (see Chapter 14) cAt the time of blood sample collection for serum Br monitoring PB, phenobarbital; LEV, levetiracetam; Br, bromide; NA, not available
Principles of Anti-epileptic Treatment
significant difference in the mean seizure frequency per month during treatment between IMP and PB groups, however the percentage of seizure-free dogs during treatment was higher in the PB group (SPC, 2013). Dogs with status epilepticus were excluded from this study. There are no comparative double-blinded, randomized, parallel clinical trials on efficacy of other new generation AEMs used as initial monotherapy. When comparing results of separate studies, zonisamide monotherapy was less effective than PB monotherapy in decreasing seizure frequency ³50% of baseline (Boothe et al., 2012; Chung et al, 2012). Felbamate monotherapy was very effective in decreasing seizure frequency in dogs with focal seizures (Ruehlmann et al., 2001). FBM use is not recommended in animals with hepatic dysfunction.
Bromide is generally the first choice AEM in dogs with hepatic dysfunction or in dogs with concurrent disorders requiring lifelong administration of potentially hepatotoxic medications. However, if costs are not a limiting factor, monotherapy with levetiracetam may represent a valid alternative to Br in this patient group. In addition, LEV may be used as adjunctive AEM to Br for the initial 3–4 months of Br treatment (until Br reaches steady-state reference range), particularly in dogs with high seizure frequency or severity. LEV can be used in cats, whereas Br use is currently not recommended in cats due to the potential for life-threatening respiratory adverse effects.
Adjunctive anti-epileptic treatment
Adjunctive anti-epileptic treatment should be started when excessive seizure activity
(e.g. seizure frequency and/or severity) persists despite steady-state serum concentrations of the AEM within the high reference range or in case of persistent unacceptable adverse effects. It is important to use any AEM correctly before considering the animal pharmacoresistant to it and adding on further AEMs. See definitions of pharmacoresistant epilepsy in Chapter 2. In animals on PB monotherapy and inadequate seizure control or persistent unacceptable adverse effects, serum PB concentrations must be checked and if appropriate the dosage (dose and or dosing interval) should be adjusted to improve efficacy and tolerability (see Chapter 13).
When treating dogs in the UK resistant to PB and with normal hepatic function the authors generally follow the Royal College of Veterinary Surgeons guidelines and add on Br or IMP (as second line AEM); subsequently, with further poor control, IMP or Br or LEV (as third) is added, then ZNS (as fourth or fifth), and then GBP or PGB (as fifth or sixth) if necessary (Fig. 12.1). However, a different order of adjunctive AEMs may be preferred based on numerous variables including animal’s seizure type, frequency and aetiology, co-morbidities and co-medications, as well as the pet owner’s lifestyle and financial circumstances. Where not limited by specifically using veterinary licensed AEMs, the authors recommend considering LEV or ZNS as second choice medications when PB is not satisfactorily effective. In dogs that do not respond to monotherapy with IMP, if hepatic function is normal, PB should be considered as first adjunctive or alternative therapy, whereas Br and/or LEV should be used in dogs with hepatic disease. If no improvement is observed after addition of an AEM (once steady state has been reached and maintained for adequate time) the ineffective AEM can be gradually substituted for an alternative AEM. In people requiring AEM polytherapy, data suggest that if seizure freedom is achievable, it is most likely to occur after addition of the third AEM, and with AEM substitution rather than multiple additions. However, a few patients can become seizure free on their fourth, fifth, sixth or even seventh AEM (Stephen and Brodie, 2012).
Intermittent adjunctive treatment with LEV or clorazepate (0.5–2.0 mg/kg Po tid) for a couple of days may be used in dogs with predictable (e.g. the owner can recognize the prodromal signs) seizures or with cluster seizures (see Chapters 16, 21 and 23). Combinations of AEMs should be carefully selected based on potential for synergy that is not associated with unfavourable pharmacokinetic interactions and toxicity (Table 12.3). Further studies are needed in veterinary medicine to identify optimal AEM combinations.
In cats resistant to PB, LEV may represent a safe and effective second AEM of choice (Fig. 12.2). Data on safety and efficacy of GBP and ZNS are currently limited in cats; however, these as well as other new AEMs may have a role in feline seizure management in the future. Before the advent of new AEMs, DZM was considered the second AEM of choice for epileptic cats; however, its use has fallen out of favour due to concerns of acute hepatic necrosis (see Chapter 21).
Alternative anti-epileptic treatment
Alternative anti-epileptic treatment should be instituted when discontinuation of the initial AEM is necessary due to life-threatening adverse reactions. The choice of alternative AEM is affected by type of adverse reaction
(e.g. in dogs Br and/or LEV will be the preferred AEM in case of hepatotoxicity; IMP may be preferred in case of blood dyscrasias or dermatologic adverse effects) as well as all the other variables (Box 12.1). The alternative AEM type and dosage (e.g. loading versus maintenance) should be chosen in order to achieve steady state in the reference range as rapidly as possible (Tables 12.6 and 12.7) to minimize the risk of breakthrough seizures following rapid discontinuation of the previous AEM. IMP, LEV, GBP and PGB reach steady state in 1–2 days, ZNS in approximately 3-4 days, whereas Br administered at maintenance dosage requires approximately 3 months. A loading dosage would allow reaching steady state more rapidly than maintenance dosage, however this is associated with pronounced adverse effects (particularly for Br) (see Chapter 14).
Alternative anti-epileptic mono- or adjunctive therapy can also be considered when an AEM is poorly tolerated at low to moderate dose or is ineffective (Stephen and Brodie, 2012).
Efficacy of AEMs
The majority of studies on efficacy of AEMs in veterinary medicine are open label rather than randomized blinded placebo controlled and sample size is often modest. A decrease in seizure frequency following placebo administration has been described in epileptic dogs in three randomized controlled trials (Muñana et al., 2010). Epilepsy is a waxing and waning disorder with changes in seizure frequency over time. Initial monotherapy or adjunctive anti-epileptic treatment is more likely to be initiated when seizure frequency increases and subsequent short-term decrease in seizure frequency may be due to the natural course of the disease rather than the effect of the AEM. Long term follow-ups (e.g. at least 3 times the longest interictal interval before AEM initiation), standardized outcome measures and whenever possible, a placebo control group would help to more accurately evaluate the efficacy of anti-epileptic treatment. In addition, studies on efficacy of AEMs should be interpreted based on whether the investigated canine population originates from a referral or first opinion institution, as the severity of epilepsy is likely to be greater in dogs referred to a neurologist than in dogs initially seen in first-opinion practice. Direct comparison among results of various studies is limited due to variability in inclusion criteria, seizure severity, AEM dosage, outcome measures, duration and modality of assessment of seizure frequency before and after treatment initiation (e.g. retrospective versus prospective). Comparison of efficacy of different AEMs should be done based on prospective double-blinded, randomized, parallel clinical trials. A parallel-designed clinical trial compares the results of two different treatments on two separate groups of patients.
Choice of Anti-epileptic Medication Dosage
The dosage (dose and dosing interval) of AEM needs to be tailored to the individual. Recommended AEM dosages are based on population pharmacokinetic studies predominantly in healthy animals and, when available, on serum or plasma reference ranges. AEM pharmacokinetics may differ in epileptic versus healthy animals as well as among breeds and individuals of the same species due to variability in absorption, distribution, metabolism and excretion. Reference serum concentration ranges may not apply to all individuals and are unknown for new generation AEMs in dogs and cats.
In general, unless the animal has frequent and severe seizures, anti-epileptic treatment can be started at the lower end of the recommended maintenance dose (Tables 12.6 and 12.7) and increased gradually based on efficacy, tolerability and serum concentration monitoring for first generation AEMs (Box 12.2). This approach helps to identify the lowest effective AEM dose and serum concentration in the individual and allows the body to gradually adapt to the medication minimizing occurrence and severity of adverse effects.
Alternatively, animals with frequent and severe seizures should be administered a loading dose although this often results in more pronounced adverse effects than with initial routine maintenance dose (particularly with Br). The amount of the loading dose necessary to achieve steady state increases proportionately with the half-life of the AEM to be loaded. Loading doses and formulas to calculate them based on desired serum AEM concentration and AEM pharmacokinetic parameters have been detailed in AEM-specific chapters (see Chapters 13, 14 and 16).
AEM that induce their own metabolism, such as PB in dogs, require dose and sometimes also dosing interval adjustment over time to maintain adequate serum concentration and seizure control (see Chapter 13) (Box 12.2). With renally excreted AEM, dosage reduction should be considered in patients with impaired renal function and this could result in decreased AEM renal clearance, subsequently increased AEM concentrations and toxicity. Therapeutic monitoring is an important tool in guiding dosage adjustment in these cases. Gradual AEM dosage reduction should be considered in animals with satisfactory seizure control and persistent adverse effects (e.g. sedation and ataxia).
The calculation of new daily dose of AEM based on current and desired serum concentrations is:
AEM total daily dosage (in mg) = (desired serum AEM concentration/actual serum AEM concentration) × actual AEM total daily dosage (in mg) (12.1)
Dosage modulation may also be required when combining AEMs or when AEMs are used concurrently to other medications resulting in pharmacokinetic interactions (Table 12.3). For example, PB alters disposition of ZNS in dogs and people requiring a dose increase of ZNS.
optimal dose and dosing interval for several new generation AEMs has not been definitively established in dogs and cats.
Box 12.2. Indications for therapeutic monitoring of AEM concentrations.
Therapeutic Monitoring of AEMs and Treatment Modulation
Therapeutic monitoring of AEMs is the measurement for clinical use of AEM (and/or AEM active metabolite) concentrations in body fluids, usually serum or plasma (Johannessen and Johannessen Landmark, 2008). The clinical value of therapeutic monitoring rests on the assumptions that clinical effects of AEMs correlate better with AEM concentrations than with dose and that the AEM concentration in the sampled body fluid (e.g. blood) is highly correlated with the AEM concentration at the receptor sites in the brain. Therefore the relative value of therapeutic monitoring depends on the characteristics of the AEM (Patsalos et al., 2008). Therapeutic monitoring of first generation AEMs such as PB and Br is an important tool in anti-epileptic treatment individualization and optimization because first-generation AEMs have narrow therapeutic ranges and significant inter-individual variability in their pharmacokinetics (absorption, distribution, metabolism and excretion). Therapeutic monitoring of new generation AEMs is not routinely performed and it may be of limited value when there is no established correlation between serum AEM concentration and therapeutic efficacy or toxicity. Data from randomized controlled trials on the clinical benefits of AEM therapeutic monitoring in humans is limited. However, non-randomized trials and long-lasting clinical experience suggest that therapeutic monitoring of both first and new generation AEMs has a valuable role in guiding patient management if used appropriately (Patsalos et al., 2008).
Knowledge of serum or plasma AEM concentrations, combined with information on occurrence of seizures and AEM adverse effects can help to individualize AEM dosage, optimize seizure control while avoiding or minimizing adverse effects and prevent toxicity. Therapeutic monitoring can also help to assess compliance and to guide dosage adjustment in case of:
(e.g. autoinduction of PB metabolism in dogs, addition or discontinuation of an interacting medication, change in AEM formulation, or change in dietary chloride content in dogs on Br).
General guidelines for therapeutic monitoring are listed in Box 12.2. AEM-specific therapeutic monitoring indications are detailed in the respective chapter (see Chapters 13–22).
Reference ranges and individualization of AEM dosage
Terms such as target ranges, therapeutic ranges, effective ranges, optimal ranges, reference ranges and reference concentrations have been variably used in the human and veterinary AEM literature, either interchangeably or with different meanings (Patsalos et al., 2008). In this book we will use the following definitions based on the ILAE recommendations for therapeutic monitoring of AEMs (Patsalos et al., 2008) (Boxes 12.3 and 12.4).
Box 12.3. Definition of reference range (Patsalos et al., 2008).
The reference range can be defined as a range of drug concentrations, which is quoted by a laboratory and specifies a lower limit below which a therapeutic response is relatively unlikely to occur, and an upper limit above which toxicity is relatively likely to occur. Optimal clinical response is expected in most patients with serum AEM concentration within the reference range.
Box 12.4. Definition of individual therapeutic range (Patsalos et al., 2008).
The individual therapeutic range can be defined as the range of drug concentrations that is associated with the best achievable response
(e.g. seizure freedom with good tolerability, or the best compromise between improvement in seizure control and concentration-related adverse effects) in an individual. Therefore this range will vary in different individuals. However, the therapeutic range of many individuals will lie within, or at least close to, the reference range, if the reference range originated from reliable research.
Reference ranges for PB and Br are well established (Tables 12.6 and 12.7); however, individual animals may respond therapeutically below the range or adversely within the range. Therefore combining information on seizure control and presence of AEM adverse effects with AEM serum concentration over time can help to identify the individual therapeutic concentration and modulate treatment accordingly. In humans, it has been demonstrated that reference ranges of PB and other AEMs may vary with different types of seizures as well as severity of epilepsy (Schmidt et al., 1986). It is unknown whether this is also the case in veterinary medicine, however, identification of the individual therapeutic range could address this situation.
Reference ranges of new generation AEM are overall unknown in dogs and cats and not fully established in humans, however the proposed range for humans is often used as guideline in veterinary medicine (Tables 12.6 and 12.7). Therapeutic monitoring may help to individualize treatment even in the absence of defined reference ranges, by utilization of the concept of ‘individual therapeutic concentrations’ based on intra-individual comparisons of AEM serum concentrations and associated clinical response (Johannessen and Tomson, 2006; Patsalos et al., 2008).
The individual therapeutic concentration should be established during a period of satisfactory seizure control and no undesired effects. Ideally, it should be based on two separate determinations obtained several weeks to months apart rather than on a single determination. Identification of the individual therapeutic concentration can help to identify potential causes of therapeutic failure and optimize patient management during further follow up. For example, in case of breakthrough seizures after a period of seizure control, the actual serum AEM concentration can be compared with the previously established individual therapeutic concentration to determine if a drop in concentration played a role. If a decrease in serum AEM concentration has occurred, the clinician should identify its cause by considering owner compliance, drug interactions or changes in AEM disposition (e.g. increased Br excretion due to increased chloride intake or AEM formulation change).
The AEM dosage should be adjusted in order to re-establish the previous serum AEM concentration resulting in optimal effect (see equation
12.1 on p 364). If the serum AEM concentration is unchanged, but it can be safely increased, the dose and, if necessary, also the dosing interval are modified accordingly. Alternatively, if the serum AEM concentration is in the high reference range and seizure control remains unsatisfactory or unacceptable adverse effects persist, adjunctive therapy should be considered.
In addition, in animals with mild types of epilepsy, the individual therapeutic concentration may be below the lower limit of the reference range. Therefore a dosage increase of AEM is not necessary in animals with prolonged seizure freedom in spite of serum AEM concentrations below the lower limit of the reference range.
one limitation of the individual therapeutic concentration approach is that changes (e.g. progression or regression) in the underlying seizure disorder may require establishment of a new individual therapeutic concentration over time (Krasowski, 2010).
Timing of therapeutic monitoring
Therapeutic monitoring should be performed when the AEM has reached steady state after initiation of anti-epileptic treatment or a change in AEM dosage (Tables 12.6 and 12.7). Time to steady state (Tss) depends on the AEM’s elimination half-life, dosage (maintenance versus loading) and kinetics. When the AEM is initiated at maintenance dosage, steady state is achieved in approximately five half-lives under conditions of first-order kinetics in a one-compartment distribution model
(e.g. the medication is rapidly and evenly distributed throughout the body). When the AEM is administered at loading dosage, serum concentration monitoring should be performed the day after completion of loading and one half-life (of that AEM) later to ensure that the maintenance dosage is adequate to maintain the desired concentration. In general, clinical response to anti-epileptic therapy should not be evaluated until steady state has been reached, however seizure control may be achieved earlier in some individuals. If therapeutic monitoring is undertaken before reaching steady state following a dose increment, the steady-state serum concentration at that dose will be underestimated. Consequently, if a further dose increase is undertaken, this may eventually result in toxicity for the patient.
The time elapsed between the last administered AEM dose and blood sample collection should be consistent to allow comparison of results, particularly for AEMs with a short half-life (£12 h). Collection of a blood sample immediately before the next oral dose (trough) would allow consistency of sampling as well as assessment of the lowest concentration (Cmin) in the dosing interval. For some AEMs, such as LEV, time of the day (e.g. morning versus afternoon) should be consistent as diurnal variation in LEV excretion may occur (Moore et al., 2010). For AEMs with a long half-life, such as Br, the fluctuation in serum concentration is negligible (during a dosing interval) after steady state has been reached, therefore sampling can be performed at any time. When toxicity is suspected or overdosing has occurred, sampling should be performed at peak AEM (Cmax) as well as trough (Cmin) concentration.
Assessment of steady-state serum AEM peak (Cmax) and trough (Cmin) concentrations can be used to determine AEM half-life and therefore to evaluate the need to alter the dosing interval in AEM with short half-life or when a decrease in PB half-life is suspected (i.e. loss of seizure control in a dog on prolonged PB treatment) (see example in Box 12.5).
Food may prolong absorption of some AEM and delay time to peak concentration therefore it is preferable to take peak serum AEM samples on fasted animals. Serum separation blood collection tubes containing a clot activator may falsely decrease serum PB concentration and therefore only standard blood collection tubes should be used (Boothe et al., 1996).
Therapeutic monitoring and change of AEM formulation
Differences in bioavailability between brand and generic AEM formulations may affect steady-state concentrations and potentially seizure control, therefore therapeutic monitoring
Box 12.5. Example of AEM half-life determination based on therapeutic monitoring and subsequent AEM dosage adjustment.
Signalment: 6-year 8-month-old, male neutered Italian spinone
Diagnosis: Idiopathic epilepsy
Treatment: Phenobarbitone for the previous 2.5 years, with gradual dose increase over time based on therapeutic and clinical monitoring. Most recent dosage: 8 mg/kg every 12 h
Presenting complaint: Partial loss of seizure control and episodes of sedation and ataxia (approximately 4 h after PB administration)
General physical and neurologic examination: normal
Investigations: Haematology and serum biochemistry: no significant abnormalities
Steady-state serum PB peak (Cmax) concentration = 35 mg/ml and trough (Cmin) concentration = 24 mg/ml
Elimination half-life = 0.693/Kel = 0.693/0.05 = 13.86 h
Kel = ln (peak PB serum concentration/trough PB serum concentration)/T2−T1 = ln1.5/8 = 0.4/8 = 0.05
where Kel = elimination rate constant;
ln = natural logarithm;
T1 = time interval in hours between administration of PB and collection of peak sample (this should be about 4 h);
T2 = Time interval in hours between administration of PB and collection of trough sample (this should ideally be as close as possible to 12 h).
The PB dosage was changed to 5 mg/kg every 8 h; 3 weeks later serum PB peak (Cmax) concentration =
28.8 mg/ml and trough (Cmin) concentration = 27.2 mg/ml; sedation and ataxia resolved and seizure control improved.
Box 12.6. Key points for successful anti-epileptic treatment.
should be performed before and after the formulation change to identify potential clinically significant alterations in steady-state AEM concentrations (Perucca et al., 2006; Karalis et al., 2014). Sampling time must be consistent to allow this comparison. When patients are switched to a formulation with modified-release characteristics (e.g. from an immediate-release to a sustained-release formulation), or when dosing schedule is changed (for example, from every 12 h to every 8 h administration), interpretation of therapeutic monitoring results should also take into account the expected variation in diurnal AEM concentration profile. Collection of two or more blood samples at different intervals after AEM administration may be desirable to fully assess the concentration profile change (Patsalos et al., 2008).
Therapeutic monitoring and pharmacokinetic interactions
Therapeutic monitoring should be performed any time clinically relevant pharmacokinetic interactions are expected. Pharmacokinetic interactions involve a change in the absorption, distribution, metabolism or elimination of the affected medication. Pharmacokinetic interactions can occur between AEMs, as well as between AEMs and various medications, including antimicrobials and herbal medicines (see Table 12.3 and Chapter 25). The most prevalent pharmacokinetic interactions are those associated with induction or inhibition of enzymes involved in AEM metabolism. An increase in the rate of AEM metabolism will lead to a decrease in its serum concentrations and possibly reduction in therapeutic response. A decrease in the rate of AEM metabolism will lead to an increase in its serum concentrations and potentially toxicity. When a pharmacokinetic interaction is anticipated, AEM concentration measurement should be performed before adding the new medication, in order to establish a baseline. Further measurements should be taken at appropriate times (e.g. steady state, loss of seizure control, suspected toxicity) after the potentially interacting agent has been added in order to assess the need for a dose adjustment (Patsalos and Perucca, 2003).
Therapeutic monitoring and pathological states
Several pathological states including renal failure, hepatic failure, infectious diseases, surgery and illnesses severe enough to warrant intensive care (such as head trauma) can alter physiology to a degree that AEM concentrations can be affected. In addition, the medications used to treat the pathological state can cause interactions that also affect AEM concentrations (Patsalos et al., 2008). Therefore AEM concentrations should be monitored in individuals that develop various illnesses that can alter AEM absorption, metabolism, clearance and protein binding, so that AEM dosage can be adjusted accordingly in order to maintain seizure control and prevent toxicity.
Salivary therapeutic monitoring
In humans, saliva has been used as an alternative medium to serum or plasma for therapeutic monitoring of AEMs. Salivary therapeutic monitoring allows noninvasive, simple and easily repeatable sampling, and reflects the pharmacologically active concentration in serum of many AEMs. Salivary therapeutic monitoring has proven to be useful for several AEMs including gabapentin, lacosamide, levetiracetam, phenobarbital, phenytoin, primidone, topiramate and zonisamide (Patsalos and Berry, 2013).
When Can Anti-epileptic Treatment be Discontinued?
Generally treatment for idiopathic epilepsy involves lifelong AEM administration. However, remission with or without medication has been reported in dogs and cats with epilepsy. In a study of Danish Labrador Retrievers, 24% of dogs were classed as being in remission
(e.g. seizure free for two years); with only 1 (6%) of these receiving antiepileptic treatment (Berendt et al., 2002). In a further Danish study of 63 dogs with epilepsy, the remission rate (both spontaneous remission and remission with treatment) was 15% (Berendt et al., 2007). In a Swiss study of Labrador Retrievers, 30% of dogs treated with PB became seizure free, with an average follow-up period of
4.8 years (Heynold et al., 1997).
In selected cases with seizure freedom for 1 to 2 years, or longer (depending on previous seizure frequency), gradual tapering of AEM over 6 months or longer could be considered. The pet owner must be aware that seizures may recur anytime during AEM dose reduction or after discontinuation. However, some individuals may remain seizure free. In a study including 36 idiopathic epileptic cats, seizures recurred in six out of eight (86%) cats in which PB was reduced or discontinued after 1 seizure-free year, however two (24%) cats remained seizure free for more than 2 years after PB discontinuation (Pakozdy et al., 2012). If seizures recur, retreatment should be reinstituted promptly.
In humans with prolonged seizure remission (generally 2 or more years), the decision to discontinue anti-epileptic treatment is done on an individual basis considering relative risks and benefits. Individuals with the highest probability of remaining seizure-free are those who had no structural brain lesions, a short duration of epilepsy, few seizures before pharmacologic control, and AEM mono-therapy (o’Dell and Shinnar, 2001; Shih and ochoa, 2009). Seizure relapse rates of 12% to 67% have been reported in AEM withdrawal human trials (Shih and ochoa, 2009). Unfortunately, lack of information on risk factors associated with seizure relapse in animals makes it impossible to predict which veterinary patients can be successfully weaned off AEMs. In addition, it is unknown how difficult it would be to re-establish seizure control in dogs and cats following AEM withdrawal and seizure relapse. In humans, reinstitution of anti-epileptic treatment after seizure recurrence was efficacious in 64–91% of patients with a mean follow-up of 1–9 years. Chronic AEM-resistant epilepsy developed in up to 23% of patients with a recurrence. Factors associated with poor treatment outcome of treating recurrences were symptomatic aetiology, focal epilepsy and cognitive deficits (Schmidt and Löscher, 2005).
Pet Owner Education
optimal communication and education (especially if repeated over time) of the pet’s owner is the best way to promote compliance and successful management of an epileptic pet. Pet owners who are inadequately educated may change the AEM dosage, medicate irregularly, discontinue AEM or change veterinarian repeatedly as they have unrealistically high expectations of treatment results, as well as anxiety about AEM-induced adverse effects or potential organ damage. The owners of an epileptic pet need to be educated thoroughly on:
diet changes and concurrent conditions and medications can also be added;
References
Adsumalli, V.E., Gilchirst, J.R., Wichmann, J.K., Kucharczyk, N. and Sofia, R.D. (1992) Pharmacokinetics of felbamate in pediatric and adult beagle dogs. Epilepsia 33, 955–960.
Al-Tahan, F. and Frey, H.H. (1985) Absorption kinetics and bioavailability of phenobarbital after oral administration to dogs. Journal of Veterinary Pharmacology and Therapeutics 8, 205–207.
Bailey, K.S., Dewey, C.W., Boothe, D.M., Barone, G. and Kortz, G.D. (2008) Levetiracetam as an adjunct to Phenobarbital treatment in cats with suspected idiopathic epilepsy. Journal of the American Veterinary Medical Association 232, 867–872.
Benedetti, M.S., Coupez, R., Whomsley, R., Nicolas, J.M., Collart, P. and Baltes, E. (2004) Comparative pharmacokinetics and metabolism of levetiracetam, a new anti-epileptic agent, in mouse, rat, rabbit and dog. Xenobiotica 34, 281–300.
Berendt, M., Gredal, H., Pedersen, L.G., Alban, L., Alving, J. (2002) A cross-sectional study of epilepsy in Danish Labrador Retrievers: prevalence and selected risk factors. Journal of Veterinary Internal Medicine 16, 262–268.
Berendt, M., Gredal, H., Ersbøll, A.K., Alving, J. (2007) Premature death, risk factors, and life patterns in dogs with epilepsy. Journal of Veterinary Internal Medicine 21, 754–759.
Boothe, D.M. and Perkins, J. (2008) Disposition and safety of zonisamide after intravenous and oral single dose and oral multiple dosing in normal hound dogs. Journal of Veterinary Pharmacology and Therapeutics 31, 544–553.
Boothe, D.M., Simpson, G. and Foster, T. (1996) Effects of serum separation tubes on serum benzodiazepine and phenobarbital concentrations in clinically normal and epileptic dogs. American Journal of Veterinary Research 57, 1299–1303.
Boothe, D.M., George, K.L. and Couch, P. (2002) Disposition and clinical use of Bromide in cats. Journal of the American Veterinary Medical Association 221, 1131–1135.
Boothe, D.M., Dewey, C. and Carpenter, D.M. (2012) Comparison of phenobarbital with bromide as a first-choice antiepileptic drug for treatment of epilepsy in dogs. Journal of the American Veterinary Medical Association 240, 1073–1083.
Boynosky, N.A. and Stokking, L.B. (2013) Potassium bromide-associated panniculitis. Journal of Small Animal Practice Sep 6. doi: 10.1111/jsap.12129. [Epub ahead of print]
Carnes, M.B., Axlund, T.W. and Boothe, D.M. (2011) Pharmacokinetics of levetiracetam after oral and intravenous administration of a single dose to clinically normal cats. American Journal of Veterinary Research 72, 1247–1252.
Chang, Y., Mellor, D.J. and Anderson, T.J. (2006) Idiopathic epilepsy in dogs: owners’ perspectives on management with phenobarbitone and/or potassium bromide. Journal of Small Animal Practice 47(10), 574–581.
Chung, J.Y., Hwang, C.Y., Chae, J.S., Ahn, J.O., Kim, T.H., Seo, K.W., Lee, S.Y. and Youn, H.Y. (2012) Zonisamide monotherapy for idiopathic epilepsy in dogs. New Zealand Veterinary Journal 60(6), 357–359.
Cochrane, S.M., Black, W.D., Parent, J.M., Allen, D.G. and Lumsden, J.H. (1990a) Pharmacokinetics of phenobarbital in the cat following intravenous and oral administration. Canadian Journal of Veterinary Research 54, 132–138.
Cochrane, S.M., Parent, J.M., Black, W.D., Allen, D.G. and Lumsden, J.H. (1990b) Pharmacokinetics of phenobarbital in the cat following multiple oral administrations. Canadian Journal of Veterinary Research 54, 309–312.
Cotler, S., Gustafson, J.H. and Colburn, W.A. (1984) Pharmacokinetics of diazepam and nordiazepam in the cat. Journal of Pharmaceutical Sciences 73, 348–351.
Dewey, C.W., Guiliano, R., Boothe, D.M., Berg, J.M., Kortz, J.D., Joseph, R.J. and Budsberg, S.C. (2004) Zonisamide Therapy for Refractory Idiopathic Epilepsy in Dogs. Journal of the American Animal Hospital Association 40, 285–291.
Dewey, C.W., Bailey, K.S., Boothe, D.M., Badgley, B.L. and Cruz-Espindola, C. (2008) Pharmacokinetics of single-dose intravenous levetiracetam administration in normal dogs. Journal of Veterinary Emergency and Critical Care 18, 153–157.
Dewey, C.W., Cerda-Gonzalez, S. and Levine, J.M. (2009) Pregabalin as an adjunct to phenobarbital, potassium bromide, or a combination of phenobarbital and potassium bromide for treatment of dogs with suspected idiopathic epilepsy. Journal of American Veterinary Medical Association 235, 1442–1449.
Finnerty, K.E., Barnes Heller, H.L., Mercier, M.N., Giovanella, C.J., Lau,V.W. and Rylander, H. (2014) Evaluation of therapeutic phenobarbital concentrations and application of a classification system for seizures in cats: 30 cases (2004–2013). Journal of American Veterinary Medical Association 244(2), 195–199. doi: 10.2460/javma.244.2.195.
Glauser, T., Ben-Menachem, E., Bourgeois, B., Cnaan, A., Guerreiro, C., Kälviäinen, R., Mattson, R., French, J.A., Perucca, E., Tomson, T.; ILAE Subcommission on AED Guidelines (2013) Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 54(3), 551–563.
Govendir, M., Perkins, M. and Malik, R. (2005) Improving seizure control in dogs with refractory epilepsy using gabapentin as an adjunctive agent. Australian Veterinary Journal 83, 602–608.
Hasegawa, D., Kobayashi, M., Kuwabara, T., Ohmura, T., Fujita, M. and Orima, H. (2008) Pharmacokinetics and toxicity of zonisamide in cats. Journal of Feline Medicine and Surgery 10, 418–421.
Heynold, Y., Faissler, D., Steffen, F. and Jaggy, A. (1997) Clinical, epidemiological and treatment results of idiopathic epilepsy in 54 Labrador retrievers: a long term study. Journal of Small Animal Practice 38, 7–14.
Johannessen, S.I. and Johannessen Landmark, C. (2008) Value of therapeutic monitoring in epilepsy. Expert Reviews of Neurotherapy 8(6), 929–939.
Johannessen, S.I. and Tomson, T. (2006) Pharmacokinetic variability of newer antiepileptic drugs: when is monitoring needed? Clinical Pharmacokinetics 45(11), 1061–1075.
Johannessen Landmark, C. and Patsalos, P.N. (2010) Drug interactions involving the new second- and third-generation antiepileptic drugs. Expert Review of Neurotherapeutics 10(1), 119–140.
Kaminski, R.M., Rogawski, M.A., Klitgaard, H. (2014) The potential of antiseizure drugs and agents that act on novel molecular targets as antiepileptogenic treatments. Neurotherapeutics 11(2), 385–400.
Karalis, V., Macheras, P., Bialer, M. (2014) Generic products of antiepileptic drugs: a perspective on bioequivalence, bioavailability, and formulation switches using Monte Carlo simulations. CNS Drugs 28(1), 69–77.
Kiviranta, A.M., Laitinen-Vapaavuori, O., Hielm-Björkman, A. and Jokinen, T. (2013) Topiramate as an add-on antiepileptic drug in treating refractory canine idiopathic epilepsy. Journal of Small Animal Practice 54(10), 512–520.
Krasowski, M.D. (2010) Therapeutic Drug Monitoring of the Newer Anti-Epilepsy Medications. Pharmaceuticals (Basel) 3(6), 1909–1935.
KuKanich, B. and Cohen, R.L. (2011) Pharmacokinetics of oral gabapentin in greyhound dogs. Veterinary Journal 187,133–135.
Martinez, S.E., Bowen, K.A., Remsberg, C.M., Takemoto, J.K., Wright, H.M., Chen-Allen, A.V. and Davies,
N.M. (2012) High-performance liquid chromatographic analysis of lacosamide in canine serum using ultraviolet detection: application to pre-clinical pharmacokinetics in dogs. Biomedical Chromatography 26(5), 606–609.
Moore, S.A., Muñana, K.R., Papich, M.G. and Nettifee-Osborne, J. (2010) Levetiracetam pharmacokinetics in healthy dogs following oral administration of single and multiple doses. American Journal of Veterinary Research 71, 337–341.
Muñana, K.R. (2013) Update: seizure management in small animal practice. Veterinary Clinics of North America Small Animal Practice 43(5), 1127–1147.
Muñana, K.R., Zhang, D. and Patterson, E.E. (2010) Placebo Effect in Canine Epilepsy Trials. Journal of Veterinary Internal Medicine 24(1), 166–170.
Muñana, K.R., Thomas, W.B., Inzana, K.D., Nettifee-Osborne, J.A., McLucas, K.J., Olby, N.J., Mariani, C.J. and Early, P.J. (2012) Evaluation of levetiracetam as adjunctive treatment for refractory canine epilepsy: a randomized, placebo-controlled, crossover trial. Journal of Veterinary Internal Medicine 26, 341–348.
Musicco, M., Beghi, E., Solari, A. and Viani, F. (1997) Treatment of first tonic-clonic seizure does not improve the prognosis of epilepsy. First Seizure Trial Group (FIRST Group). Neurology 49(4), 991–998.
O’Dell, C. and Shinnar, S. (2001) Initiation and discontinuation of antiepileptic drugs. Neurologic Clinics 19, 289–311.
Orito, K., Saito, M., Fukunaga, K., Matsuo, E., Takikawa, S., Muto, M., Mishima, K., Egashira, N. and Fujiwara,
M. (2008) Pharmacokinetics of zonisamide and drug interaction with phenobarbital in dogs. Journal of Veterinary Pharmacology and Therapeutics 31, 259–264.
Pakozdy, A., Asghar, S.A., Michael, L., Alexander, T.G., Halasz, P. and Thalhammer, J.G. (2012) Treatment and long-term follow-up of cats with suspected primary epilepsy. Journal of Feline Medicine and Surgery Oct 22.
Patsalos, P.N. and Berry, D.J. (2013) Therapeutic drug monitoring of antiepileptic drugs by use of saliva. Therapeutic Drug Monitoring 35(1), 4–29.
Patsalos, P.N. and Perucca, E. (2003) Clinically important drug interactions in epilepsy: general features and interactions between antiepileptic drugs. Lancet Neurology 2, 347–356.
Patsalos, P.N., Berry, D.J., Bourgeois, B.F., Cloyd, J.C., Glauser, T.A., Johannessen, S.I., Leppik, I.E., Tomson, T. and Perucca, E. (2008) Antiepileptic drugs – best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 49(7), 1239–1276.
Patterson, E.E., Goel, V., Cloyd, J.C., O’Brien, T.D., Fisher, J.E., Dunn, A.W. and Leppik, I.E. (2008) Intramuscular, intravenous and oral levetiracetam in dogs: safety and pharmacokinetics. Journal of Veterinary Pharmacology and Therapeutics 31, 253–258.
Pedersoli, W.M., Wike, J.S. and Ravis, W.R. (1987) Pharmacokinetics of single doses of phenobarbital given intravenously and orally to dogs. American Journal of Veterinary Research 48, 679–683.
Perucca, E., Albani, F., Capovilla, G., Bernardina, B.D., Michelucci, R. and Zaccara, G. (2006) Recommendations of the Italian League against Epilepsy working group on generic products of antiepileptic drugs. Epilepsia 47(Suppl. 5), 16–20.
Peters, R.K., Schubert, T., Clemmons, R., Vickroy, T. (2014) Levetiracetam Rectal Administration in Healthy Dogs. Journal of Veterinary Internal Medicine. Jan 13. doi: 10.1111/jvim.12269.
Platt, S.R., Adams, V., Garosi, L.S., Abramson, C.J., Penderis, J., De Stefani, A. and Matiasek, L. (2006) Treatment with gabapentin of 11 dogs with refractory idiopathic epilepsy. Veterinary Record 159, 881–884.
Podell M. (2013) Seizures. In: Platt, S. and Olby, N. (eds) BSAVA Manual of Canine and Feline Neurology, 4th edn. BSAVA, Gloucester, UK, pp. 117–135.
Podell, M. and Fenner, W.R. (1993) Bromide therapy in refractory canine idiopathic epilepsy. Journal of Veterinary Internal Medicine 7, 318–327.
Radulovic, L.L., Türck, D., Von Hodenberg, A., Vollmer, K.O., McNally, W.P., DeHart, P.D., Hanson, B.J., Bockbrader, H.N. and Chang, T. (1995) Disposition of gabapentin (neurontin) in mice, rats, dogs, and monkeys. Drug Metabolism and Disposition 23, 441–448.
Ravis, W.R., Pedersoli, W.M. and Wike, J.S. (1989) Pharmacokinetics of phenobarbital in dogs given multiple doses. American Journal of Veterinary Research 50, 1343–1347.
Rieck, S., Rundfeldt, C. and Tipold, A. (2006) Anticonvulsant activity and tolerance of ELB138 in dogs with epilepsy: a clinical pilot study. Veterinary Journal 172(1), 86–95.
Ruehlmann, D., Podell, M. and March, P. (2001) Treatment of partial seizures and seizure-like activity with felbamate in six dogs. Journal of Small Animal Practice 42, 403–408.
Salazar, V., Dewey, C.W., Schwark, W., Gleed, R.D., Horne, W. and Ludder, J.W. (2009) Pharmacokinetics of single-dose oral pregabalin administration in normal dogs. Veterinary Anaesthesia and Analgesia 36, 574–580.
Schmidt, D. and Löscher, W. (2005) Uncontrolled epilepsy following discontinuation of antiepileptic drugs in seizure-free patients: a review of current clinical experience. Acta Neurologica Scandinavica 111(5), 291–300.
Schmidt, D., Einicke, I. and Haenel, F.T. (1986) The influence of seizure type on the efficacy of plasma concentrations of phenytoin, phenobarbital, and carbamazepine. Archives of Neurology 43, 263–265.
Schwartz-Porsche, D., Jurgens, U., May, T., Gerhardt, M., Boenigk, H.E. and Krebs, B. (1990) Pharmacokinetics of bromide and bromide therapy in canine epilepsy. Proceedings of the 4th Annual European Society of Veterinary Neurology, Bern, Switzerland, pp. 32–34.
Shih, J.J. and Ochoa, J.G. (2009) A systematic review of antiepileptic drug initiation and withdrawal. Neurologist 15(3), 122–131.
Siao, K.T., Pypendop, B.H. and Ilkiw, J.E. (2010) Pharmacokinetics of gabapentin in cats. American Journal of Veterinary Research 71, 817–821.
Stephen, L.J. and Brodie, M.J. (2009) Selection of antiepileptic drugs in adults. Neurologic Clinics 27(4), 967–992.
Stephen, L.J. and Brodie, M.J. (2012) Antiepileptic drug monotherapy versus polytherapy: pursuing seizure freedom and tolerability in adults. Current Opinion in Neurology 25(2), 164–172.
Streeter, A.J., Stahle, P.L., Holland, M.L., Pritchard, J.F. and Takacs, A.R. (1995) Pharmacokinetics and bioavailability of topiramate in the beagle dog. Drug Metabolism and Disposition, 23, 90–93.
Summary of product characteristics (SPC) (2013) Imepitoin Summary of product characteristics. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/veterinary/ 002543/WC500140840.pdf (accessed 19/9/13).
Trepanier, L.A. and Babish, B.J. (1995) Pharmacokinetics properties of Bromide in dogs after the intravenous and oral administration of single doses. Research in Veterinary Science 58, 248–251.
Trepanier, L.A., Van Schoick, A., Schwark, W.S. and Carrillo, J. (1998) Therapeutic serum drug concentrations in epileptic dogs treated with potassium Bromide alone or in combination with other anticonvulsants: 122 cases (1992–1996). Journal of the American Veterinary Medical Association 213, 1449–1453.
Von Klopmann, T., Rambeck, B. and Tipold, A. (2007) Prospective study of Zonisamide therapy for refractory idiopathic epilepsy in dogs. Journal of Small Animal Practice 48, 134–138.
Volk, H., Coleing, J.A.L., Platt, S.R. and Chandler, K. (2007) Clinical presentation and response to treatment of cats with epilepsy. In: Proceedings of British Small Animal Veterinary Association Congress, pp. 487–488.
Volk, H.A., Matiasek, L.A., Luján Feliu-Pascual, A., Platt, S.R. and Chandler, K.E. (2008) The efficacy and tolerability of levetiracetam in pharmacoresistant epileptic dogs. Veterinary Journal 176, 310–319.
Wessmann, A., Volk, H.A., Parkin, T., Ortega, M. and Anderson, T.J. (2011) Living with canine idiopathic epilepsy: a questionnaire-based evaluation of quality of life. Proceedings of ECVN symposium 2011, Trier, Germany.
Wright, H.M., Chen, A.V., Martinez, S.E. and Davies, N.M. (2012) Pharmacokinetics of oral rufinamide in dogs. Journal of Veterinary Pharmacology and Therapeutics 35, 529–533.
13 Phenobarbital
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Phenobarbital (PB) (5-ethyl-5-phenylbarbituric acid) (Fig. 13.1) is a potent anti-epileptic medication (AEM) whose properties were first described in 1912 (Hauptmann, 1912). PB is efficacious in the treatment of different seizure types. It is available in both oral and injectable preparations. PB is licensed for veterinary use in epileptic dogs in the UK. Various oral preparations (including 15, 30, 60 mg tablets) are available in different countries.
PB is commonly the first medication of choice in epileptic dogs and cats due to its pharmacokinetic profile, relative safety, affordable cost, efficacy and greater data from veterinary studies compared to other AEMs. The preferential use of PB, compared with bromide, as first choice AEM in epileptic dogs is supported by a recent study (Boothe et al., 2012).
In humans, PB is still used for neonatal and childhood seizures, for drug-resistant convulsive and non-convulsive status epilepticus as well as for various seizure types in low-and middle-income countries (Brodie and Kwan, 2012). In high-income countries, PB use has declined due to the availability of other AEMs with fewer adverse effects.
Mechanism of Action
PB increases seizure threshold and decreases the spread of discharge to surrounding neurons (Boothe, 2012a). The exact mechanism of action is not completely understood. The primary mechanism involves enhancement of postsynaptic neuronal inhibition by increasing responsiveness to the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) in the CNS. Phenobarbital binds at the GABAA receptor to both directly activate GABA receptor- gated chloride channels and to increase the affinity of GABA for its own receptor by allosteric effect (Fig. 13.2). This results in opening of the chloride ion channel for a longer time, greater chloride intracellular influx and concentration, which produces hyperpolarization of the resting membrane potential of the post-synaptic neuron (Ticku and Kulkarni, 1992). Other proposed mechanisms include interaction with glutamate receptors to decrease neuronal excitatory postsynaptic currents (Nardou et al., 2011), inhibition of voltage-gated calcium channels resulting in decreased calcium influx into neurons and competitive binding with the picrotoxin site of the chloride channel.
Metabolism and Pharmacokinetics
PB has high bioavailability (86–97%) and is absorbed within 2 h after oral administration in dogs (Al-Tahan and Frey, 1985; Pedersoli et al., 1987; Ravis et al., 1989). Administration with food results in an approximate 10% decrease in extent of absorption compared to administration on an empty stomach (Thurman et al., 1990). Maximal plasma concentration is reached approximately 4 h after oral administration in dogs (Ravis et al., 1989). PB is less lipid-soluble than other barbiturates and half-life to equilibrium between plasma and CSF concentration is 16 min after IV administration of PB in dogs (Frey et al., 1979). Plasma protein binding is approximately 45–46% in dogs (Frey et al., 1979). The volume of distribution following single PB administration is 0.7 ± 0.15 l/kg in dogs and 0.77 ± 0.02 l/kg in cats (Pedersoli et al., 1987; Cochrane et al., 1990a). PB crosses the placenta and can be teratogenic. PB is metabolized primarily by hepatic microsomal enzymes and approximately 25% is excreted unchanged renally. There is individual variability in PB
O HN O HN O

absorption, excretion and elimination half-life. Pharmacokinetic parameters of PB after intravenous and oral administration in healthy dogs and cats in different studies are presented in Tables 13.1 and 13.2, respectively. Differences among studies may be related to population as well as methodology variations.
The elimination half-life after a single oral administration of approximately 5 mg/kg of PB has been reported as 46.3 ± 11.3 h (Thurman et al., 1990) or 88.7 ± 19.6 h (Ravis et al., 1989) in dogs and 47.6 ± 2.89 h in cats (Cochrane, 1990a). Steady-state concentration is reached approximately 10–20 days after initiation of oral administration at maintenance dosage. In cats administered PB at 5 mg/kg/day for 21 days, steady state was present on day 19 (although no sampling occurred between days 7 and 19 of the study), average serum PB concentration at steady state was approximately 16.4 mg/ml, Cmax was 19.3 mg/ml and Cmin was 13.4 mg/ml (Cochrane et al., 1990b).
In dogs, PB is a potent inducer of hepatic microsomal cytochrome P450 (CYP450) enzymes (Hojo et al., 2002). At clinical dosage, induction of the CYP subfamilies CYP1A, CYP2B, CYP2C and CYP3A has been demonstrated

Fig. 13.2. Neuronal receptor targets for phenobarbital.
Pharmacokinetic parameters
| Reference | Dose administered | F (%) | Vd/F (l/kg) | Cl (ml/h/kg) | Clt/F (ml/h/kg) | Cmax (µg/ml) | Tmax (h) | T½ (h) | MRT (h) | |
| Al-Tahan and | 10 mg/kg IV once | Mean | NA | NA | NA | NA | NA | NA | 56 | NA |
| Frey, 1985 | Median | NA | NA | NA | NA | NA | NA | 56 | NA | |
| Min | NA | NA | NA | NA | NA | NA | 42 | NA | ||
| Max | NA | NA | NA | NA | NA | NA | 72 | NA | ||
| 10 mg/kg PO once | Mean | 91 | NA | NA | NA | 15.3 | 6 | 52 | NA | |
| Median | 91 | NA | NA | NA | 15 | 6 | 52 | NA | ||
| Min | 86 | NA | NA | NA | 14 | 4 | 44 | NA | ||
| Max | 96 | NA | NA | NA | 17 | 8 | 62 | NA | ||
| Pedersoli | 5.5 mg/kg PO once | Mean ± SD | 88.1 ± 12.4 | NA | NA | NA | NA | NA | NA | NA |
| et al., 1987 | 15 mg/kg PO once | Mean ± SD | 96.8 ± 9.0 | NA | NA | NA | NA | NA | NA | NA |
| 5.5 mg/kg IV once | Mean ± SD | NA | 0.7 ± 0.15 | 5.60 ± 2.31 | NA | NA | NA | 92.6 ± 23.7 | 124 ± 34.3 | |
| 15 mg/kg IV once | Mean ± SD | NA | 0.69 ± 0.14 | 6.66 ± 0.78 | NA | NA | NA | 72.3 ± 15.5 | 106 ± 23.4 | |
| Ravis | 5.5 mg/kg PO once | Mean ± SD | NA | 706 ± 129 | NA | 5.58 ± 1.89 | 8.94 ± 1.67 | 3.80 ± 1.48 | 88.7 ± 19.6 | 120 ± 22 |
| et al., 1989 | 5.5 mg/kg/day | Mean ± SD | NA | 720 ± 175 | NA | 10.2 ± 1.7 | 27.69 ± 4.04 | 4.60 ± 0.89 | 47.3 ± 10.7 | 73 ± 13 |
| PO for 90 days | ||||||||||
| 15 mg/kg PO once | Mean ± SD | NA | 736 ± 107 | NA | 7.28 ± 1.07 | 22.24 ± 2.83 | 4.40 ± 1.52 | 99.6 ± 22.6 | 107 ± 27 | |
| 11 mg/kg/day | Mean ± SD | NA | 712 ± 144 | NA | 15.6 ± 2.53 | 40.50 ± 4.12 | 2.80 ± 1.48 | 31.1 ± 4.4 | 47.4 ± 7.9 | |
| PO for 90 days | ||||||||||
| Thurman | 5 mg/kg PO once | Mean ± SD | NA | NA | NA | NA | 6.5 ± 0.66 | 2–4 | 46.3 ± 11.3 | NA |
| et al., 1990 | 5 mg/kg PO | Mean ± SD | NA | NA | NA | 13.3 ± 1.6 | 18.68 ± 1.54 | 2–4 | 29.3 ± 4.6 | NA |
| for 22 days |
376 L. De Risio
PO, per os; IV, intravenously; F, bioavailability; Vd, volume of distribution; Cl, clearance; Vd/F, volume of distribution/extent of absorption; Clt/F, total body clearance/extent of absorption; C, serum maximum concentration; T, time to maximum serum concentration; T½, elimination half-life; MRT, mean residence time
maxmax
Pharmacokinetic parameters
| Reference | Dose administered | F (%) | Vd (l/kg) | Cl (ml/h/kg) | AUC (h.µg/ml) | Tmax (h) | T½ (h) | |
| Cochrane et al., 1990a | 10 mg/kg IV once | Mean ± SD | NA | 0.93 ± 0.04 | 11.3 ± 0.76 | 918.9 ± 69.3 | NA | 58.8 ± 4.21 |
| Min | NA | 0.78 | 7.68 | 656.9 | NA | 41.3 | ||
| Max | NA | 1.14 | 14.4 | 1301.5 | NA | 77 | ||
| 10 mg/kg PO once | Mean ± SD | 120 ± 12 | 0.73 ± 0.04 | 6.97 ± 0.6 | 1518.1 ± 139.3 | NA | 76 ± 6.96 | |
| Min | 82 | 0.6 | 4.7 | 1151.7 | 1 | 46.2 | ||
| Max | 179 | 0.9 | 9.1 | 2145.2 | 1.5 | 96.3 | ||
| Cochrane et al., 1990b | 5 mg/kg PO once | Mean ± SD | NA | 0.77 ± 0.02 | 11.4 ± 0.44 | NA | NA | 47.6 ± 2.89 |
| Min | NA | 0.7 | 9.23 | NA | NA | 40.8 | ||
| Max | NA | 0.87 | 12.5 | NA | NA | 57.8 | ||
| 5 mg/kg/day PO | Mean ± SD | NA | 0.70 ± 0.04 | 11.3 ± 0.78 | NA | NA | 43.3 ± 2.92 | |
| for 21 days | Min | NA | 0.63 | 7.88 | NA | NA | 35 | |
| Max | NA | 0.93 | 14.6 | NA | NA | 56.3 |
PO, per os; IV, intravenously; F, bioavailability; Vd, volume of distribution; Cl, clearance; Vd/F, volume of distribution/extent of absorption; Tmax, time to maximum serum concentration; T½, elimination half-life
Phenobarbital 377
(Graham et al., 2002; Hojo et al., 2002). Chronic administration in dogs can thus lead to increased clearance of hepatically metabolized medications (including PB itself) as well as endogenous compounds (such as thyroid hormones) (Gieger et al., 2000; Müller et al., 2000a; Hojo et al., 2002). As a result, with chronic PB administration, its total body clearance increases and elimination half-life decreases progressively (in most dogs). Total body clearance and mean elimination half-life were
5.58 ± 1.89 ml/h/kg and 88.7 ± 19.6 h, respectively, after a single PB administration of 5.5 mg/ kg; these values changed to 10.2 ± 1.7 ml/h/ kg and 47.3 ± 10.7 h, respectively, after PB administration of 5.5 mg/kg/day for 90 days (Ravis et al., 1989). In another study, mean elimination half-life at 6 months following initiation of PB (46 ± 31 h 95% CI, 29 to 56 h) was significantly less than that at baseline (68 ± 31 h; 95% CI, 56 to 88 h). The greatest decrease in half-life in a single dog was from 120 h on day 8 to 21 h at study end (month 6) and the shortest half-life measured in any dog was 14 h at study end (Boothe et al., 2012). This decrease in elimination half-life can result in reduction of PB serum concentrations, significant changes in peak and trough concentrations during a single dosing interval and therapeutic failure. Therefore monitoring of serum PB concentrations is very important for dosage modulation over time.
Hepatic enzyme induction does not seem to occur in the cat and therefore chronic PB administration should not alter steady-state concentration in individual cats. This may be due to low activities of CYP2C in cats (Shah et al., 2007). In one study, elimination half-life was 47.6 ± 2.9 following a single oral administration of 5 mg/kg/day of PB and 43.3 ± 2.9 after 21 days of treatment at the same dosage (Cochrane et al., 1990b). Also other pharmacokinetic parameters did not vary significantly following chronic administration (Cochrane et al., 1990b). However, significant inter-individual differences in elimination kinetics have been identified in cats and therefore serum PB monitoring should be used to individualize PB dosage (Cochrane et al., 1990b).
PB metabolism can also be affected by the diet and urine pH (Maguire et al., 2000; Fukunaga et al., 2008). Dogs fed a low-protein or low-protein and low-fat diet, have a more rapid elimination of PB (Maguire et al., 2000). Similarly, urine alkalinization (PH >7.5) increases renal excretion of PB (Fukunaga et al., 2008).
Pharmacokinetic Interactions and Adverse Reactions
In dogs, chronic PB administration can affect the disposition of other co-administered medications which are metabolized by CPY450 subfamilies CYP1A, 2B, 2C and 3A and/or bound to plasma a1-acid glycoprotein (AGP) (Hojo et al., 2002). AGP is an important plasma protein involved in the binding and transport of many drugs, especially basic (alkaline) compounds, as well as endogenous substances (Huang, 2012). Therefore by increasing plasma AGP, PB can affect the unbound fraction of other concurrently administered medications (Hojo et al., 2002).
PB can alter the pharmacokinetics and as a consequence may decrease the therapeutic effect of other AEMs including benzodiazepines, levetiracetam and zonisamide (Forrester et al., 1993; Wagner et al., 1998; Orito et al., 2008; Moore et al., 2011) as well as corticosteroids, cyclosporine, metronidazole, voriconazole, digoxin, digitoxin, phenylbutazone and some anaesthetics (e.g. thiopental). These interactions have not been reported in cats and are unlikely to occur due to the apparent lack of hepatic CYP induction.
Pharmacokinetic interaction with benzodiazepines
Oral administration of PB at 2.5 mg/kg every 12 h for 30 days, resulted in a significant decrease in total benzodiazepine (diazepam, nordiazepam and oxazepam) maximal plasma concentration, area under the curve and mean residence time in dogs administered 2 mg/kg of diazepam rectally. The area under the curve was significantly decreased post-PB treatment compared to pre-PB treatment in dogs administered 2 mg/kg of diazepam IV (Wagner et al., 1998).
After oral administration of PB (5 mg/ kg, q12h) and clorazepate (2 mg/kg, q12h) for 44 consecutive days, maximal nordiazepam concentrations were significantly lower, ranging from 209.6 to 698.5 ng/ml (mean,
399.3 ± 155.6 ng/ml) than after single oral administration of clorazepate (2 mg/kg), ranging from 569.6 to 1387.9 ng/ml (mean, 880.2 ±
248.9 ng/ml). Mean area under the curve (AUC) on day 1 (mean, 3.37 ± 0.598 ng/min/ml) was significantly greater than AUC on day 44
(1.66 ± 0.308 ng/min/ml). Oral clearance was significantly greater on day 44 (12.44 ±
2.55 ml/min/kg) compared with that on day 1 (6.16 ± 1.35 ml/min/kg). Values for area under the first moment curve, oral volume of distribution, mean residence time, and elimination half-life were not significantly altered by concurrent administration of PB (Forrester et al., 1993). The increased clearance and decreased plasma concentrations of benzodiazepines were attributed to PB induction of CYP (Forrester et al., 1993; Wagner et al., 1998).
Pharmacokinetic interaction with levetiracetam
Administration of PB at 2.0–3.3 mg/kg orally every 12 h for 21 days significantly altered the pharmacokinetics of concurrently administered levetiracetam (Moore et al., 2011). Compared with values determined when levetiracetam was the sole AEM administered, concurrent administration of PB resulted in a decrease in levetiracetam peak concentration from 32.39 ± 6.76 to 18.22 ± 8.97 (P = 0.0071), a decrease in elimination half-life from 3.43 ± 0.47 to 1.73 ± 0.22, and an increase in oral clearance from 124.93 ± 26.93 to 252.99 ± 135.43 ml/h/kg. The extent of these pharmacokinetic changes varied among individuals. It has been speculated that the increased disposition of levetiracetam is caused by PB induction of oxidative enzymes in dogs. Therefore, the levetiracetam oral dosage may need to be adjusted
(i.e. up to 60 mg/kg every 8 h) when concurrently administered with PB (Moore et al., 2011).
Pharmacokinetic interaction with zonisamide
Repeated oral administration of PB at 5 mg/ kg every 12 h for 30–35 days decreased the maximum serum concentration, apparent elimination half-life and bioavailability of zonisamide and increased its clearance. The enhanced clearance of zonisamide lasted at least 10 weeks after the discontinuation of PB (Orito et al., 2008). This pharmacokinetic interaction could be the result of PB induction of CYP3A activity and plasma AGP (Orito et al., 2008). A larger dosage of zonisamide (e.g. 10 mg/kg every 12 h) is required when administered concurrently with PB.
Pharmacokinetic interaction with CYP450 inhibitors
Concurrent administration of PB and medications that inhibit hepatic microsomal CYP450 enzymes such as cimetidine, omeprazole, lansoprazole, chloramphenicol, trimethoprim, fluoroquinolones, tetracycline, ketoconazole, fluconazole, itraconazole, fluoxetine, felbamate (see Chapter 18) and topiramate (see Chapter 19) may inhibit PB metabolism, increase serum concentration and result in toxicity (Boothe, 2012b). Silybin and silymarin, major constituents of the milk thistle, which is used in several hepatic support preparations, have been shown to inhibit a variety of CYP450 enzymes such as CYP3A4, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP2E1 and also major hepatic glucuronosyltransferases in vitro (Sridar et al., 2004; Hermann and von Richter, 2012). This inhibition could affect metabolism of PB. However, it is unknown if this effect is clinically relevant at commonly recommended doses in dogs.
Other pharmacokinetic interactions
Concurrent administration of PB can impair the absorption of griseofulvin and therefore decrease plasma concentration and efficacy of this anti-fungal medication (Jamali and Axelson, 1978).
Activated charcoal impairs the absorption of PB from the gastrointestinal tract. Administration of ammonium chloride for urine alkalinization increases renal elimination of PB and consequently decreases serum PB concentrations (Fukunaga et al., 2008).
Transient and Dose-Related Adverse Effects of PB in Dogs and Cats
Common adverse effects of PB in dogs and cats include:
These adverse effects are dose dependent, generally occur in the first and second week of treatment or dosage increase and usually subside in the subsequent 1–3 weeks due to the development of pharmacokinetic and pharmacodynamic tolerance (Frey, 1986; Thurman et al., 1990; Quesnel et al., 1997; Pakozdy et al., 2012; see Chapter 3). Sedation may be profound and persistent in dogs with intracranial tumours and alternative AEM to PB may be necessary. Polydipsia and polyuria are uncommon in cats compared to dogs.
Hyperexcitability, hyperactivity, restlessness and aggression can occasionally occur in dogs and tend to be transient during the first weeks of PB treatment (Farnbach, 1984; Chang et al., 2006). Hyperactivity may resolve following PB dosage increase (Farnbach, 1984). Behavioural changes such as fear/anxiety, defensive aggression and abnormal perception (e.g. barking without apparent cause, chasing shadows or light spots, aimless pacing, staring into space) can occur in dogs at the time of development of idiopathic epilepsy (Shihab et al., 2011). Therefore it may be difficult to differentiate neurobehavioural co-morbidities of epilepsy from PB (or other AEM)-related behavioural abnormalities.
Suspected Idiosyncratic Adverse Effects of PB in Dogs
Uncommonly reported adverse effects include:
A direct cause-and-effect relationship has not been proven for most of these adverse effects, however the majority of them are suspected to be idiosyncratic adverse drug reactions, which resolve following discontinuation of PB.
Hepatotoxicity
PB treatment has been associated with hepatic damage in dogs (Dayrell-Hart et al., 1991; Gaskill et al., 2005). Induction of cytochrome P450 by PB significantly increases hepatic production of reactive oxygen species, thus increasing the risk of hepatic injury (Shaik and Mehvar, 2010). It is still unclear whether PB-related hepatotoxicity occurs as a direct dose-dependent adverse effect or as an idiosyncratic reaction. PB-induced hepatic cirrhosis appears to be a dose- and treatment-duration dependent adverse effect as it has been reported in dogs administered high PB dosage (>12 mg/ kg/day) resulting in high serum PB concentrations (>35 mg/ml) for prolonged (>6 months) periods (Bunch et al., 1982; Dayrell-Hart et al., 1991). In contrast, idiosyncratic reactions are unrelated to serum PB concentrations or treatment duration and result in signs of acute hepatic failure (Dayrell-Hart et al., 1991).
Clinical signs of PB-induced hepatotoxicity include sedation, ataxia, inappetence or anorexia, icterus and ascites (in severely affected dogs) (Dayrell-Hart et al., 1991; Müller et al., 2000b). Commonly reported serum laboratory abnormalities in dogs with PB-induced hepatotoxicity include:
• elevation in serum ALT activity ³2 times the upper normal limit;
In a retrospective study of 18 dogs with PB-induced hepatotoxicity, hypoalbuminaemia occurred in 78%, and elevations in ALP, ALT, fasting bile acids, and total bilirubin were observed in 100, 83, 80 and 50% of tested dogs, respectively (Dayrell-Hart et al., 1991). The average duration of treatment was 39 months, and 72% of dogs had a serum PB trough concentration greater than 40 mg/ml. Grossly, the liver was reduced in size with extensive fibrosis interspersed with areas of nodular hyperplasia (Bunch et al., 1982; Dayrell-Hart et al., 1991).
Hepatotoxicity may be reversible if it is detected early and PB is decreased or discontinued. However, hepatic damage can sometimes be irreversible and ultimately fatal (Dayrell-Hart et al., 1991). Concurrent administration of PB and other potentially hepatotoxic medications can increase the risk of hepatotoxicity and should be avoided whenever possible (Dayrell-Hart et al., 1991; Müller et al., 2000b). Monitoring serum biochemistry (including fasted ammonia and pre- and post-prandial bile acids) as well as serum PB concentration every 3–6 months can help to minimize the risk of PB-induced hepatotoxicity in dogs on long-term PB therapy. If serum PB concentration is >35 mg/ml, the PB dose needs to be decreased and, depending on seizure control, an additional AEM (preferably not one which is hepatically metabolized) has to be initiated.
Hepatotoxicity has not been reported in cats treated with PB.
Haematologic abnormalities (cytopenias)
Anaemia and/or thrombocytopenia and/or neutropenia have been reported in dogs administered maintenance dosage of PB for 30–240 days (median 100.5 days) and in a dog with acute phenobarbital intoxication (Jacobs et al., 1998; Khoutorsky and Bruchim, 2008; Bersan et al., 2012; Habock and Pakosdy, 2012). Clinical presentation was characterized by poor appetite and lethargy in dogs administered PB at maintenance dosage. PB serum concentration ranged from 13.2 to 30.5 mg/ml (median 19 mg/ml). Discontinuation of PB resulted in resolution of the haematologic abnormalities in 2–110 days (median 17 days) in the majority of dogs (Thompson and Johnstone, 1983; Jacobs et al., 1998; Weiss, 2005; Khoutorsky and Bruchi, 2008; Bersan et al., 2012). Although the exact mechanism is unknown, most likely the cytopenias result from bone marrow toxicity rather than an immune-mediated process directed at circulating cells as both bone marrow necrosis and myelofibrosis have been associated with PB administration (Thompson and Johnstone, 1983; Weiss and Smith, 2002; Weiss, 2005).
Superficial necrolytic dermatitis
Skin disease has been reported in dogs with serum PB concentrations ranging from 22.8 to 66 mg/ml (mean maximal 43.5 ± 15.1 mg/ml), 1.7–11 years (median 6 years) after initiation of PB treatment (Bloom et al., 1992; Outerbridge et al., 2002; March et al., 2004). Superficial necrolytic dermatitis is characterized by alopecia, erythaema, crusting, exudation and ulceration of the skin. The lesions are generally distributed over the ventral aspect of the abdomen, mucocutaneous junctions (Fig. 13.3), pressure points and distal portions of the extremities. The footpads are commonly affected with crusting, hyperkeratosis and fissures (Fig 13.4). Painful footpad lesions result in inactivity, lethargy, lameness and reluctance to walk.


PB-induced superficial necrolytic dermatitis has not been associated with hepatic failure (March et al., 2004). It has been theorized that chronic PB administration leads to accelerated hepatic catabolism of amino acids, whose deficiency could be the cause of superficial necrolytic dermatitis (March et al., 2004). Prognosis overall has been reported as guarded to poor. However, prompt recognition of superficial necrolytic dermatitis, discontinuation of PB and any other medication that could adversely affect hepatic metabolism, hepatic support (diet and/or supplements), parenteral and oral administration of supplemental amino acids, zinc and essential fatty acids and treatment of the secondary skin infections (with bacteria, yeasts and/or dermatophytes) can sometimes result in a favour-able outcome.
Pancreatitis
Pancreatitis has been reported as a possible adverse effect in epileptic dogs treated with PB alone or in combination with potassium bromide (KBr) (Podell and Fenner, 1993; Hess et al., 1999; Gaskill et al., 2000). Reported prevalence is 0.3% in epileptic dogs on PB mono-therapy and at least 10% in dogs treated with PB and KBr (Gaskill and Cribb, 2000). However, a cause effect relationship has not been proven yet. It has been speculated that polyphagia secondary to PB and/or KBr could lead to dietary indiscretion and consequently to pancreatitis (Podell and Fenner, 1993). An increased risk for elevated serum canine pancreatic lipase immunoreactivity concentrations has been reported in epileptic dogs treated with PB or KBr alone or in combination (Steiner et al., 2008). Fasting hypertriglyceridaemia has been associated with high post-prandial serum triglyceride concentrations, which is considered a risk factor for pancreatitis. Fasting hypertriglyceridaemia was identified in 33% (19/57) of epileptic dogs treated with PB alone or in combination with KBr for 3 months or longer (Kluger et al., 2008); 16% of dogs had a history of pancreatitis; 27% of dogs in which canine pancreatic lipase immunoreactivity was measured had high values; 63% of epileptic dogs treated with PB alone or in combination with KBr had a body condition score ³6. Hypertriglyceridaemia was attributable to delayed clearance of chylomicrons, most likely as a result of reduced lipoprotein lipase activity or hepatic very low density lipoprotein over-production causing lipoprotein lipase saturation. No association was identified between serum triglyceride concentration and PB dosage, serum PB concentration and seizure activity. It is unclear whether fasting hypertriglyceridaemia was related to an overweight body condition, an idiosyncratic reaction to PB or a multifactorial process (Kluger et al., 2008). To minimize the risk of pancreatitis in epileptic dogs treated with PB alone or in conjunction with KBr, feeding a low-fat diet, providing regular exercise to maintain a healthy body condition score and performing periodical monitoring of fasting serum triglyceride concentration has been recommended (Kluger et al., 2008).
Dyskinesia
Dyskinesia and anxiousness have been reported in a dog receiving PB for 8 weeks (Kube et al., 2006). The dyskinesia was characterized by fine intermittent contractions of the facial muscles and ears and severe intermittent contractions of the cervical and shoulder muscles that would cause the affected dog to fall. The dyskinesia and anxiousness gradually decreased as the dosage of PB was reduced and resolved after PB was discontinued.Awide variety of dyskinesias associated with anticonvulsant administration (including PB, gabapentin and felbamate) have been reported in humans. The pathophysiology of dyskinesia associated with anticonvulsant administration is unknown.
Hypoalbuminaemia
Hypoalbuminaemia has been reported in one dog administered standard doses of PB. There was no evidence of hepatic dysfunction or protein-losing nephropathy. The dog developed oedema and ultimately required a plasma transfusion. The albumin returned to normal concentration after discontinuation of PB (Rusbridge, 2013). It has been suggested that PB may induce a defect in albumin synthesis (Gieger et al., 2000).
Suspected Idiosyncratic Adverse Effects of PB in Cats
Suspected idiosyncratic adverse effects of PB in cats include:
• severe thrombocytopenia (platelet count 20 × 109/l; reference range 190–400 × 109/l) resulting in generalized ecchymosis 5 weeks after initiation of PB treatment and 2 weeks after a PB dosage increase from 1.9 to
2.9 mg/kg every 12 h (see Chapter 3; Quesnel et al., 1997);
Each of the above adverse reactions has been reported in individual cats. A direct cause-and-effect relationship between PB and most of these adverse effects has not been proven. However, clinical signs have resolved within 1 to 2 weeks after discontinuation of PB in all cats. PB was not discontinued in the cat with transient and dexamethasoneresponsive facial pruritus.
A dose-related decrease in serum concentration of vitamin K-dependent coagulation factors II, VII and X has been reported in cats administered PB at 10–40 mg/kg/day in an experimental study (Solomon et al., 1974). Administration of vitamin K concurrently with PB corrected the coagulation factors abnormality within 1 week (Solomon et al., 1974).
Alternative AEMs in dogs and cats requiring discontinuation of PB due to life-threatening adverse effects
When PB needs to be discontinued due to a potentially life-threatening adverse effect, this should be done rapidly but cautiously to avoid withdrawal seizures and under close medical supervision.
Rapidity of PB withdrawal can range from 50% dose reduction a week over 2 weeks to 50% decrease each day for 1–3 days and subsequent discontinuation. Rapidity of PB withdrawal is affected by severity of the adverse effect, seizure control, serum concentrations of concurrent AEM (if any was administered) and time required to achieve steady-state reference concentrations of the AEM to be used as alternative to PB. Loading with an alternative AEM should be initiated promptly in order to achieve target serum concentrations before serum PB concentration decreases (Box 13.1). In animals that were already administered another AEM in addition to PB (e.g. Br) before the PB-induced life-threatening adverse effect developed, the dose of the other AEM may be adjusted (e.g. Br mini-loading, see Chapter 14) to maintain seizure control after PB is discontinued, however additional AEM is often required.
Cytoprotective agents
If acute hepatotoxicity is the reason for PB discontinuation N-acetylcysteine should be administered initially (140 mg/kg IV once, then 70 mg/kg IV q6h for seven treatments). Longer term hepatic support can be provided with s-adenosylmethionine (SAMe) administered at 20 mg/kg/day PO on an empty stomach or other hepato-protectants (Webster and Cooper, 2009b). Preparations containing silybin and silymarin should be avoided until several weeks after PB discontinuation as they may delay PB disposition (see Pharmacokinetic interaction with CYP450 inhibitors).
PB-related Laboratory Changes
Laboratory changes related to PB chronic administration in dogs include elevation in hepatic enzyme activity, hypercholesterolaemia, hypertriglyceridaemia and alterations in some endocrin function testing.
Increased hepatic enzyme activity
Administration of PB at approximately 5 mg/ kg twice daily for 27 weeks resulted in elevation of the serum activity of alkaline phosphatase (ALP) (above the reference range upper limit since week 5), alanine transaminase (ALT) (at the high end of the reference range or slightly above its upper limit) and of gamma-glutamyltransferase (GGT) (transiently and generally within the high end of the reference range) as well as transient hypoalbuminaemia (generally within
Box 13.1. Options for alternative AEMs in dogs and cats requiring discontinuation of PB due to life-threatening adverse effects.
In dogs:
• Loading with levetiracetam at 60 mg/kg PO, IM or IV once (see Chapter 16) followed by maintenance dosage at 20 mg/kg, PO, every 8 h (at least until bromide concentrations within the reference have been reached) and concurrent maintenance dosage of potassium bromide (KBr at 30–40 mg/kg once or divided twice daily) (see Chapter 14).
• Loading with KBr at 625 mg/kg divided in eight or more doses over 48 h, or 125 mg/kg/day divided in
3–4 daily administrations for 5 consecutive days (see Chapter 14 for other loading protocols and
routes of administration) followed by maintenance dosage (KBr at 30–40 mg/kg once or divided twice daily). Br loading is often associated with pronounced adverse effects (e.g. nausea, vomiting, diarrhoea, sedation, ataxia and pelvic limb weakness, polydipsia, polyuria and polyphagia) and
hospitalization of the animal is recommended.
• Imepitoin (if hepatic function is normal) at 10–30 mg/kg every 12 h (see Chapter 22). Consider addition of Levetiracetam if withdrawal seizures occur despite administration of Imepitoin at 30 mg/kg every 12 hours
• Zonisamide (if hepatic function is normal) at 5–10 mg/kg every 12 h (see Chapter 15). A 10 mg/kg dose may be required for the initial 2–3 months of treatment as PB has been shown to enhance zonisamide clearance for at least 10 weeks after the discontinuation of PB (Orito et al., 2008).
In cats:
• Levetiracetam (20 mg/kg, PO, every 8 h) (see Chapter 16).
reference range) in dogs with no clinical signs of hepatic dysfunction (Müller et al., 2000b). Serum ALT and ALP activity were significantly higher at week 27 of PB treatment than at baseline (Müller et al., 2000b). A significant linear relationship was identified between serum PB concentration and activities of ALT, ALP, GGT and glutamate dehydrogenase (GLDH), which were higher (and above reference range for ALT and ALP) in dogs with serum PB concentration above 28 mg/ml (120 mmol/l) (Aitken et al., 2003). PB treatment duration was also associated with increased activities of ALT and ALP (Aitken et al., 2003). The degree of elevation of serum ALT and ALP activity was not correlated with pre- and post-prandial bile acids tests results (Gaskill et al., 2005). Several studies suggest that the hepatic enzyme-increased activities alone reflect PB induced increased synthesis (induction) of hepatic enzymes rather than hepatocellular damage or cholestasis and are reversible 3 to 5 weeks following discontinuation of PB (Gieger et al., 2000; Gaskill et al., 2004). A more recent study, however, suggests that serum elevations of both ALP and ALT may be attributable to subclinical hepatic injury rather than to PB induction (Gaskill et al., 2005). Serum AP isoenzyme analysis does not appear to be helpful in differentiating hepatic enzyme induction from early hepatic injury (Gaskill et al., 2005). Serum bilirubin, ammonia, fasted bile acids, and ultrasonographic and histologic evaluation of the liver are not significantly affected by the presumed enzyme-inducing effect of PB in dogs and therefore can be used to more conclusively investigate possible hepatic disease (Dayrell-Hart et al., 1991; Foster et al., 2000a; Müller et al., 2000b).
Elevation of ALP or other hepatic enzymes seems rare in cats treated with PB. In a recent study including cats administered a median PB dosage of 4 mg/kg/day (range 1.5-8.6 mg/kg/ day) for a median period of 14 months, only one of 21 cats had a transient elevation in ALT activity after prolonged PB administration. The PB dosage was not reduced in this cat, and the ALT activity returned to within the reference range at a subsequent biochemical analysis (Finnerty 2014).
Hypercholesterolaemia
Administration of PB at approximately 5 mg/kg twice daily for 27 weeks resulted in a significant increase in serum cholesterol concentrations compared to baseline. Hypercholesterolaemia resolved 3–5 weeks after discontinuation of PB (Gieger et al., 2000).
Effect on thyroid function testings
Epileptic dogs treated with PB may have thyroid function test results consistent with hypothyroidism, despite not being hypothyroid. This may be due to the effects of chronic administration of PB (Gaskill et al., 1999, 2000; Gieger et al., 2000; Müller et al., 2000a; Daminet and Ferguson, 2003), timing of blood sampling in relation to seizure activity (Gaskill et al., 1999), seizure frequency, and the effect of idiopathic epilepsy itself on thyroid function testing (von Klopmann et al., 2006). PB treatment for 3 or more weeks can result in a decrease in total thyroxine (TT4) and free T4 (fT4) concentrations below normal reference concentrations, whereas thyroid-stimulating hormone (TSH) concentrations remain normal to slightly increased (Gaskill et al., 1999, 2000; Kantrowitz et al., 1999; Gieger et al., 2000; Müller et al., 2000a; Daminet and Ferguson, 2003). These changes are probably associated with PB-enhanced metabolic clearance (hepatic metabolism and biliary excretion) of T4 and resolve 1 (TT4) to 5 (fT4) weeks after discontinuation of PB (Gieger et al., 2000; Daminet and Ferguson, 2003). In one study, serum TT4 concentrations were lower when measured within 24 h of a seizure in comparison to measurements obtained more than 24 h after a seizure (Gaskill et al., 1999). However, another study found no significant difference in the TT4 concentration dependent on the time span (more or less than 24 h) between the most recent seizure and blood sampling (von Klopmann et al., 2006). Plasma TT4 concentration below reference range and normal TSH concentration have been reported in 38% of dogs with untreated idiopathic epilepsy without clinical signs of hypothyroidism or concomitant diseases (von Klopmann et al., 2006). In addition, in this study plasma TT4 concentration was lower in dogs with a short seizure-free interval than in those with seizures occurring at greater intervals (von Klopmann et al., 2006). Certain adverse effects of PB such as lethargy and weight gain are common in hypothyroid dogs. Therefore investigation of hypothyroidism in epileptic dogs treated with PB can be very challenging.
Effect on pituitary-adrenal axis and adrenal function testing
No significant effect of PB chronic administration has been identified on adenocorticotropic hormone (ACTH) stimulation or low dose dexamethasone suppression testing in the majority of dogs (Dyer et al., 1994; Chauvet et al., 1995; Foster et al., 2000b; Müller et al., 2000a). Urinary cortisol to creatinine ratios are also unaffected (Foster, 2000b). Occasionally, plasma cortisol concentrations do not suppress in individual epileptic dogs after intravenous administration of dexamethasone at 0.01 mg/kg (Chauvet et al., 1995; Foster et al., 2000b). This may be due to individual variations in dexamethasone metabolism and clearance, specificity of the test in dogs with non-adrenal illness, or to PB-induced hepatic enzyme induction resulting in accelerated dexamethasone metabolism. In one study, baseline and ACTH-stimulated plasma aldosterone concentrations were significantly increased and above the upper reference limit in the majority of dogs following PB administration for 26 or 52 weeks (Chauvet et al., 1995).
Ultrasonographic evaluation of the adrenal glands revealed no abnormalities or changes in structure, echogenicity, thickness or shape of the adrenal glands before and after 27 weeks of PB administration at approximately 5 mg/kg every 12 h (Müller et al., 2000a).
Dosing and Monitoring Recommendations
Routine initiation
Initial oral dosage of PB as maintenance therapy is:
Subsequently the oral dosage is tailored to the individual patient based on seizure control, adverse effects and serum concentration monitoring. In most dogs, due to PB-induced CYP450 enzyme induction, the dosage needs to be progressively increased over time in order to maintain the serum concentration at steady-state reference concentration (generally, a trough serum concentration of between 20 and 35 mg/ml). In some dogs PB half-life becomes shorter than 24 h after chronic treatment and an 8-h dosing interval is indicated to minimize therapeutically relevant fluctuation of serum concentrations (Levitski and Trepanier, 2000). Measurement of both peak and trough serum PB concentrations can allow for estimation of half-life and is helpful in determining the need for more frequent dosing in epileptic dogs with poor seizure control (see Chapter 12, Box 12.5). Half-life of PB can be estimated based on peak and trough serum PB concentrations and timing of blood sampling relative to time of PB administration, using the following equations (Boothe, 2012a):
Elimination half-life = 0.693/Kel (13.1)
where Kel = ln (peak PB serum concentration/ trough PB serum concentration)/T2−T1, Kel = elimination rate constant, ln = natural logarithm, T1 = time interval in hours between administration of PB and collection of peak sample (this should be about 4 h) and T2 = time interval in hours between administration of PB and collection of trough sample (this should ideally be as close as possible to 12 h).
Immature animals, particularly kittens, may metabolize PB faster than adults, therefore serum concentration monitoring and dose adjustment as the animal grows may be necessary to prevent toxicity or treatment failure (see Chapter 3; Quesnel et al., 1997).
In addition, as PB metabolism can be affected by the diet and body composition, the dose must be re-evaluated if a dog’s diet, body weight or body composition changes during treatment (Maguire et al., 2000).
Loading dose regimen
In animals with cluster seizures, status epilepticus or high seizure frequency, it may be
necessary to reach steady-state serum concentration as rapidly as possible and therefore PB can be administered at a loading dose of 15–20 mg/kg IV, IM or PO divided in multiple doses of 3–5 mg/kg over 24–48 h. The following equation can be used to calculate the total loading dose in mg/kg in individual animals based on desired serum PB concentration (Boothe et al., 2012) (See equation
(13.2) at the bottom of the page).
When the loading is completed, the maintenance dosage can be initiated. A ‘mini’loading dose can be used in animals already on PB maintenance dosage that require a rapid increase in serum PB concentrations due to poor seizure control. This can be calculated if the current serum PB concentration is known based on (see equation 13.3 at the bottom of the page).
Reference serum PB concentrations
Reference serum PB concentrations in dogs recommended by the authors are between 20 mg/ml and 35 mg/ml (86–150 mmol/l). Ranges between 15 and 45 mg/ml (65–194 mmol/l; Levitski and Trepanier, 2000; Boothe, 2001) have been recommended by others and most laboratories, however several epileptic dogs with serum concentrations below 20 mg/ml are not well controlled and most reported cases of hepatotoxicity have occurred in dogs with concentrations above 35 mg/ml (Dayrell-Hart et al., 1991; Müller et al., 2000b). Information on serum PB concentration reference range in cats is limited and the range varies from 10 to 35 mg/ml (43.1–150 mmol/l) in different studies (see Schwartz-Porsche and Kaiser, 1989; Cochrane et al., 1990b; Quesnel et al., 1997; Chapter 3). As a general guideline, the authors suggest reference serum PB concentrations of 15–30 mg/ml (65–129 mmol/l) in cats.
To convert PB concentration from mg/l (or mg/ml) to mmol/l the conversion factor is
4.31 (see equation 13.4 at the bottom of the page).
Reference ranges should only be used as guidelines as they result from population statistics of pharmacologic studies. Each animal can respond therapeutically or adversely at a different point in the range. Therefore, monitoring of clinical signs and serum concentrations is very important to individualize oral dosage, achieve optimal seizure control and prevent toxicity. The following formula can be used to calculate PB total daily dosage in milligrams based on desired serum PB concentration, actual serum PB concentration and actual PB total daily dosage (Podell, 2004) (see equation 13.5 at the bottom of the page).
In general, the desired serum AEM concentration for individual patients should be the lowest possible concentration associated with >50% seizure frequency reduction or eradication and absence of intolerable adverse effects.
DL = desired serum concentration × Vd/F = desired serum concentration × 0.77 (13.2)
where DL = total loading dose in mg/kg, Vd = volume of distribution (e.g. 0.7 l/kg) and F = oral bioavailability (e.g. 90%)
DML = (desired serum concentration − actual serum concentration) × Vd/F = (desired serum concentration − actual serum concentration) × 0.77 (13.3)
where DML = total mini-loading dose in mg/kg, Vd = volume of distribution (e.g. 0.7 l/kg) and F = oral bioavailability (e.g. 90%).
PB concentration in mmol/l = 4.31 × PB concentration in mg/l (13.4)
PB total daily dosage in mg = (desired serum PB concentration/actual serum PB concentration) × actual PB total daily dosage in mg (13.5)
Monitoring serum PB concentrations administration), 3-h, and 6-h serum concentrations exhibiting >30% change in concen-Serum PB concentrations should be evaluated: trations throughout the day. Another study
Timing of blood sampling for therapeutic monitoring
Recommendations on optimal timing of blood collection for serum PB concentration monitoring in dogs vary among studies. One study reported no therapeutically relevant change in serum PB concentrations, throughout a daily dosing interval in 91% (30/33) of dogs administered PB at 1 to 10.9 mg/kg/day for 3 or more weeks (Levitski and Trepanier, 2000). However, 9% (3/33) of dogs had trough (within 1 h before the next scheduled identified no significant differences between the mean serum concentrations of PB in the trough and non-trough samples from dogs receiving doses ranging from 2 mg/ kg/day to 10 mg/kg/day (Monteiro et al., 2009). However, in dogs receiving a total daily dose greater than 10 mg/kg, the mean serum concentration of PB obtained from a trough sample was significantly lower than that from a non-trough sample (Monteiro et al., 2009). One more study recommended performing serum PB concentration monitoring on a trough sample as a significant difference between peak and trough phenobarbital concentration was identified in individual dogs (Boothe et al., 2012). No studies have evaluated the effect of timing of blood sampling on serum PB concentrations in cats. However, it has been recommended to obtain a trough blood sample for PB monitoring in cats (see Chapter 3; Quesnel et al., 1997).
The authors recommend collecting a trough fasted blood sample for PB monitoring in cats and in dogs administered an oral dosage higher than 5 mg/kg BID, as well as any time it is not inconvenient for the dog owner. This would allow assessment of the lowest concentration that occurs during a dosing interval and facilitate comparison of results of serial samples by maintaining consistency in the time of blood sampling in relation to the time of PB administration. In animals with seizures that are difficult to control, both a trough (as close as possible to the next scheduled administration) and peak (4 to 6 h after PB administration) sample should be collected in order to investigate the potential role of a short elimination half-life in causing therapeutic failure.
Serum PB concentrations can be falsely elevated in dogs with serum triglyceride concentrations >11.3 mmol/l; therefore serum PB concentrations should be measured on fasted blood samples (Kluger et al., 2008). Serum separation blood collection tubes containing a clot activator may falsely decrease serum PB concentration and therefore only standard blood collection tubes should be used (Boothe et al., 1996).
If PB dosage has to be reduced or the medication can be discontinued, this should be done gradually (e.g. 10–25% every 2–4 weeks) (unless a life-threatening adverse effect has developed) as withdrawal seizures may occur.
Concurrent administration of Br
When Br is added to PB to improve seizure control and there are no PB-induced life- threatening adverse effects, PB is continued at the current dosage at least until steady-state reference serum Br concentration has been reached. Subsequently, if seizure control is satisfactory but side effects (e.g. sedation, ataxia, pelvic limb weakness, polydipsia, polyuria or polyphagia) persist, PB dosage may be decreased by 10–25% every 4–6 weeks while the dog is closely monitored clinically.
When PB is used in combination with Br the serum concentration reference range may be lower than with PB monotherapy. In one study on epileptic dogs administered PB and KBr, the mean PB serum concentration was 23 mg/ml in dogs with improved seizure control following initiation of KBr. In addition, approximately 30% of dogs treated with PB and KBr maintained adequate seizure control with PB serum concentrations <15 mg/ml (Trepanier et al., 1998).
Change in PB formulation
In human medicine, loss of seizure control has been reported in association with switching between bioequivalent brand name and generic antiepileptic medications. Therefore there is the concern that different formulations of the same antiepileptic medication may not be therapeutically equivalent (Berg et al, 2008a, b; Karalis et al, 2013).
In Europe, following approval of Phenoleptil® for treatment of canine epilepsy, many dogs that were previously treated with human Luminal® tablets were switched to Phenoleptil®. Since then, there have been several anecdotal reports by clinicians and epileptic dog owners that switching from Luminal® (human tablets) to Phenoleptil®, was associated with seizure recurrence in dogs that were controlled by treatment with Luminal® (Bankstahl et al, 2013). The tablet formulation Luminal® for humans is identical with the formulation of Luminal® vet. A recent study has compared the bioavailability of PB after single oral dose administration of Luminal® vet versus Phenoleptil® in 8 healthy beagles (Bankstahl et al, 2013). Overall, the two formulations did not differ significantly with respect to pharmacokinetic parameters when mean group parameters were compared, however one dog had clearly higher peak plasma levels of PB after administration of Luminal® vet versus Phenoleptil®. Individual dogs may exhibit lower plasma levels with Phenoleptil® that could be clinically relevant (Bankstahl et al, 2013).
Efficacy
The reported efficacy of PB monotherapy in epileptic dogs and cats varies among studies. Differences may be due to inconsistency in inclusion criteria, efficacy outcome measures and follow-up duration. Overall, an efficacy of 60–93% in reducing or eradicating seizure activity has been reported in dogs and cats with serum PB concentrations within the reference range (Farnbach, 1984; Schwartz-Porsche et al., 1985; Morton and Honhold, 1988; Quesnel et al., 1997; Volk et al., 2007; Boothe et al., 2012; Pakozdy et al., 2013; Finnerty 2014).
In a recent double-blinded, randomized, parallel, clinical trial in dogs, PB treatment resulted in a significant decrease in seizure number, duration and severity and an increased seizure interval at study end (6 months) compared with baseline (Boothe et al., 2012). When the efficacy of PB was compared with that of KBr, PB treatment resulted in eradication of seizures (i.e. zero seizures at study end) (17/20 (85%) dogs) significantly more often than KBr (12/23 (52%) dogs); additionally, there was a greater percentage decrease in seizure duration (88 ± 34%), compared with KBr (49 ± 75%). PB was also better tolerated than KBr during the first 6 months of treatment. In dogs with seizure eradication, mean ± SD serum PB concentration was 25 ± 6 mg/ml (PB dosage, 4.1 ±
1.1 mg/kg; 1.9 ± 0.5 mg/lb), orally, every 12 h (Boothe et al., 2012). In this study the correlation between PB dosage and either serum drug concentrations or treatment response was poor (Boothe et al., 2012).
There is limited information on owner’s perception of epileptic dog quality of life during PB treatment. In one small study, the majority of owners thought that prolonged (>6 months) PB treatment had minimal adverse effects on the overall quality of life of their dogs and themselves (Lord and Podell, 1999). Of the owners, 74% thought that their dog’s attitude was as good as before PB treatment and 68% of owners thought that their dog’s activity level was as good as before PB treatment (Lord and Podell, 1999).
A recent study including 36 epileptic cats (with no identified underlying seizure aetiology) with a minimum follow up of 1 year reported seizure eradication in approximately 45% of cats, good (1–5 seizures/year) to moderate (6–10 seizures/year) seizure control in about 25% of cats and poor (>10 seizures/ year) seizure control in 30% of cats following PB treatment. Four of these 36 cats were also administered diazepam, gabapentin or levetiracetam (Pakozdy et al., 2013). Similar results (44% seizure free, 31% adequate seizure control, 25% inadequate seizure control) were reported in another study including 16 epileptic cats (Volk et al., 2007). In the larger study, seizure duration and severity (based on owner assessment) decreased in 72% and 69% of cats, respectively. Quality of life was considered good by the owners in 72% of cats. Results of statistical analysis suggested that early initiation of PB treatment was associated with a more favourable outcome than delayed treatment. Cats that achieved seizure eradication for over 1 year remained seizure free for a period of years, unless PB dosage was reduced or discontinued. Seizures recurred in six out of eight (86%) cats in which PB was reduced or discontinued after one seizure-free year (Pakozdy et al., 2013). In another recent study including 11 cats with structural epilepsy and 19 cats with idiopathic or probably structural epilepsy, a ³50% reduction in seizure number was achieved in 93% (28/30) of cats with a serum PB concentration between 15 and 45 mg/ml (Finnerty 2014). In the remaining 2 cats, seizure control was achieved but at serum PB concentrations of 8.0 mg/ml and
12.1 mg/ml, respectively. Thirteen (43%) of cats became seizure free after initiation of PB. Median follow-up period duration after PB treatment initiation was 14 months (range
0.63 to 91 months) for all cats (Finnerty 2014).
Response to PB and anti-epileptic treatment in general may vary among canine breeds and within individuals. Mutations in the ABCB1 gene (also called multidrug resistance 1 (MDR1) gene), may influence response to PB as well as other AEMs which are P-glycoprotein substrates (Kennerly et al., 2009; Alves et al., 2011). ABCB1 encodes for the permeability-glycoprotein (P-glycoprotein), which is an ATP-dependent trans-membrane efflux transporter in the blood-brain barrier (BBB). Differential expression of the ABCB1 gene may result in dysfunction of P-glycoprotein and lead to either drug sensitivity by increasing concentration of xenobiotics in the brain or drug resistance by limiting PB or other medication penetration of the BBB and reaching its intended site of action. The ABCB1-1D mutation (4 base-pair deletion in exon 4 of the ABCB1 gene [c.296_299del]) has been demonstrated to be the cause of increased sensitivity to ivermectin neurotoxicity in collies and other herding breed dogs (Mealey et al., 2001; Mealey, 2004). An association between ABCB1-1D mutation-related genotype and seizure outcome has been reported in epileptic collies (Muñana et al., 2012). Dogs homozygous for the ABCB1-1D mutation (M/M genotype) had a lower seizure frequency and incidence of cluster seizures, required fewer AEMs, and had a lower incidence of adverse events than dogs with either the heterozygous (M/N) or wild-type (N/N) genotype. This suggests that epileptic collies with the M/M genotype are less likely to have AEM-resistant epilepsy when compared with epileptic collies with the M/N or N/N genotype. However, this may be due to intrinsic variations in seizure severity among phenotypes rather than differences in P-glycoprotein function resulting in higher AEM concentrations in the brain of M/M dogs (Muñana et al., 2012). Another study including idiopathic epileptic Border collies reported a very low incidence (0.4%) of the ABCB1-1D mutation and identified a total of 23 variations in the ABCB1 gene: four in exons and 19 in introns. The G-allele of the c.-6-180T4G variation in intron 1 was significantly more frequent in idiopathic epileptic Border collies resistant to PB treatment than in idiopathic epileptic border collies responsive to PB (Alves, 2011). The authors hypothesized that regulatory mutations might affect the expression concentration of ABCB1 and P-glycoprotein, which could in turn influence the response to PB. Recently the frequency of the mutant G-allele has been reported as 24.9% in a population of 472 healthy Border collies in Japan (Mizukami et al, 2013). Several other genes may affect responsiveness to PB (see Chapter 2; Kennerly et al., 2009). Further studies are needed before a genetic test can be used to help predict PB response in idiopathic epileptic dogs.
Concentration of neurotransmitters in the CSF, which may reflect concentration in the CNS, has been investigated as a predictor of response to PB. Lower pretreatment CSF GABA concentrations have been correlated with a reduced response to PB in epileptic dogs. No correlation was identified between efficacy of PB and CSF glutamate concentrations (Podell and Hadjiconstantinou, 1999).
Animals are considered resistant to PB treatment when excessive seizure activity or unacceptable adverse effects (such as sedation, ataxia, polyphagia with weight gain, and polydipsia/polyuria) persist in the presence of serum concentrations which are maintained in the high reference range (30–35 mg/ml) for adequate amount of time depending on interictal period duration (see Chapter 2). In these animals, a second AEM should be initiated while PB is continued. The PB dosage can sometimes be decreased after serum reference concentrations of the second AEM have been reached without compromising seizure control. KBr (see Chapter 14) is the most commonly used add-on medication in PB-resistant dogs. Approximately 50% to 70% of PB-resistant dogs administered KBr as an adjunctive therapy experience at least a 50% reduction in seizure frequency and a decrease in seizure severity and intensity (Podell and Fenner, 1993; Trepanier et al., 1998). In addition, reduction or discontinuation of PB has been reported in 53% of dogs after the addition of Br therapy, with maintained or improved seizure control (Trepanier et al., 1998). Other AEMs (see Chapters 15 to 22) may be used as adjunctive treatment to PB or to PB and KBr. Pharmacokinetic interactions between PB and other AEMs have to be considered when choosing the medication and its dosage.
In PB refractory cats, levetiracetam (see Chapter 16) may represent a safe and effective adjunctive or alternative treatment option. Diazepam (see Chapter 21) has been used in epileptic cats in the past, however its use has fallen out of favour due to safety concerns (see Chapter 21). Further studies on safety and efficacy are needed before zonisamide (see Chapter 15) and gabapentin (Chapter 17) can be recommended in cats.
Summary Recommendations
(e.g. at least 3 times the longest interictal period before PB initiation). Asecond AEM should be administered in these animals.
References
Aitken, M.M., Hall, E., Scott, L., Davot, J.L. and Allen, W.M. (2003) Liver-related biochemical changes in the serum of dogs being treated with phenobarbitone. TheVeterinary Record 153, 13–16.
Al-Tahan, F. and Frey, H.H. (1985) Absorption kinetics and bioavailability of phenobarbital after oral administration to dogs. Journal of Veterinary Pharmacology and Therapeutics 8, 205–207.
Alves, L., Hülsmeyer, V., Jaggy, A., Fischer, A., Leeb, T. and Drögemüller, M. (2011) Polymorphisms in the ABCB1 gene in phenobarbital responsive and resistant idiopathic epileptic Border Collies. Journal of Veterinary Internal Medicine 25, 484–489.
Baho, J.M., Hostutler, R., Fenner, W. and Corn, S. (2011) Suspected phenobarbital-induced pseudolymphoma in a cat. Journal of the American Veterinary Medical Association 238, 353–355.
Bankstahl, M., Bankstahl, J.P. and Löscher, W. (2013) Is switching from brand name to generic formulations of phenobarbital associated with loss of antiepileptic efficacy?: a pharmacokinetic study with two oral formulations (Luminal(®) vet, Phenoleptil(®)) in dogs. BMC Veterinary Research 9;9:202. doi: 10.1186/1746-6148-9-202.
Berg, M.J., Gross, R.A., Tomaszewski, K.J., Zingaro, W.M. and Haskins, L.S. (2008a) Generic substitution in the treatment of epilepsy: case evidence of breakthrough seizures. Neurology 71(7), 525–530.
Berg, M.J., Gross, R.A., Haskins, L.S., Zingaro, W.M. and Tomaszewski, K.J. (2008b) Generic substitution in the treatment of epilepsy: patient and physician perceptions. Epilepsy Behaviour 13(4), 693–699. doi: 10.1016/j.yebeh.2008.06.001.
Bersan, E., Volk, H. and De Risio, L. (2012) Phenobarbital induced haematological abnormalities in 16 dogs with idiopathic epilepsy. Journal of Veterinary Internal Medicine 26, 838–839.
Bloom, P., Rosser, E. and Dunstan, R. (1992) Anti-convulsant hepatitis-induced necrolytic migratory erythema. In: Proceedings of the Second World Congress of Veterinary Dermatology, Montreal, Quebec, p. 56.
Boothe, D. (1998) Anticonvulsant therapy in small animals. Veterinary Clinics of North America - Small Animal Practice 28, 411–448.
Boothe, D.M. (2001) Therapeutic drug monitoring. In: Boothe, D.M. (ed.) Small Animal Clinical Pharmacology and Therapeutics. WB Saunders, Philadelphia, Pennsylvania.
Boothe, D.M. (2012a) Anticonvulsant and other neurologic therapies in small animals. In: Boothe, D.M. (ed.) Small Animal Clinical Pharmacology and Therapeutics, 2nd edn. Elsevier, St Louis, Missouri, pp. 950–955.
Boothe, D.M. (2012b) Introduction to drug use in dogs and cats. In: Boothe, D.M. (ed.) Small Animal Clinical Pharmacology and Therapeutics, 2nd edn. Elsevier, St Louis, Missouri, p. 62.
Boothe, D.M., Simpson, G. and Foster, T. (1996) Effects of serum separation tubes on serum benzodiazepine and phenobarbital concentrations in clinically normal and epileptic dogs. American Journal of Veterinary Research 57, 1299–1303.
Boothe, D.M., Dewey, C. and Carpenter, D.M. (2012) Comparison of phenobarbital with bromide as a first-choice antiepileptic drug for treatment of epilepsy in dogs. Journal of the American Veterinary Medical Association 240, 1073–1083.
Brodie, M.J. and Kwan, P. (2012) Current position of phenobarbital in epilepsy and its future. Epilepsia 8, 40–46.
Bunch, S.E., Castleman, W.L., Hornbuckle, W.E. and Tennant, B.C. (1982) Hepatic cirrhosis associated with long term anticonvulsivant drug therapy in dogs. Journal of the American Veterinary Medical Association 181, 357–362.
Chang, Y., Mellor, D.J. and Anderson, T.J. (2006) Idiopathic epilepsy in dogs: owners’ perspectives on management with phenobarbitone and/or potassium bromide. Journal of Small Animal Practice 47(10), 574–581.
Chauvet, A.E., Feldman, E.C. and Kass, P.H. (1995) Effects of phenobarbital administration on results of serum biochemical analyses and adrenocortical function tests in epileptic dogs. Journal of the American Veterinary Medical Association 207, 1305–1307.
Cochrane, S.M., Black, W.D., Parent, J.M., Allen, D.G. and Lumsden, J.H. (1990a) Pharmacokinetics of phenobarbital in the cat following intravenous and oral administration. Canadian Journal of Veterinary Research 54, 132–138.
Cochrane, S.M., Parent, J.M., Black, W.D., Allen, D.G. and Lumsden, J.H. (1990b) Pharmacokinetics of phenobarbital in the cat following multiple oral administration. Canadian Journal of Veterinary Research 54, 309–312.
Daminet, S. and Ferguson, D.C. (2003) Influence of drugs on thyroid function in dogs. Journal of Veterinary Internal Medicine 17, 463–472.
Dayrell-Hart, B., Steinberg, S.A., VanWinkle, T.J. and Farnbach, G.C. (1991) Hepatotoxicity of Phenobarbital in dogs: 18 cases (1985-1989). Journal of the American Veterinary Medical Association 199, 1060–1066.
Ducote, J.M., Coates, J.R., Dewey, C.W. and Kennis, R.A. (1999) Suspected hypersensitivity to phenobarbital in a cat. Journal of Feline Medicine and Surgery 2, 123–126.
Dyer, K.R., Monroe, W.E. and Forrester, S.D. (1994) Effects of short- and long term administration of phenobarbital on endogenous ACTH concentration and results of ACTH stimulation tests in dogs. Journal of the American Veterinary Medical Association 205, 315–318.
Farnbach, G.C. (1984) Serum concentrations and efficacy of phenytoin, phenobarbital and primidone in canine epilepsy. Journal of the American Veterinary Medical Association 184, 1117–1120.
Finnerty, K.E., Barnes Heller, H.L., Mercier, M.N., Giovanella, C.J., Lau, V.W. and Rylander, H. (2014) Evaluation of therapeutic phenobarbital concentrations and application of a classification system for seizures in cats: 30 cases (2004-2013). Journal of American Veterinary Medical Association 244(2), 195–199. doi: 10.2460/javma.244.2.195.
Forrester, S.D., Wilcke, J.R., Jacobson, J.D. and Dyer, K.R. (1993) Effects of a 44-day administration of phenobarbital on disposition of clorazepate in dogs. American Journal of Veterinary Research 54(7), 1136–1138.
Foster, S.F., Church, D.B. and Watson, A.D. (2000a) Effects of phenobarbitone on serum biochemical tests in dogs. Australian Veterinary Journal 78, 23–26.
Foster, S.F., Church, D.B. and Watson, A.D. (2000b) Effect of phenobarbitone on the low-dose dexamethasone suppression test and the urinary corticoid: creatinine ratio in dogs. Australian Veterinarian Journal 78, 19–23.
Frey, H.H. (1986) Use of anticonvulsants in small animals. The Veterinary Record 118, 484–486.
Frey, H.H., Gobel, W. and Loscher, W. (1979) Pharmacokinetics of primidone and its active metabolites in the dog. Archives Internationales de Pharmacodynamie et de Therapie 242, 14–30.
Fukunaga, K., Saito, M., Muto, M., Mishima, K., Fujiwara, M. and Orito, K. (2008) Effects of urine pH modification on pharmacokinetics of phenobarbital in healthy dogs. Journal of Veterinary Pharmacology and Therapeutics 31, 431–436.
Gaskill, C.L. and Cribb, A.E. (2000) Pancreatitis associated with potassium bromide/Phenobarbital combination therapy in epileptic dogs. The Canadian Veterinary Journal 41, 555–558.
Gaskill, C.L., Burton, S.A., Gelens, H.C., Ihle, S.L., Miller, J.B., Shaw, D.H., Brimacombe, M.B. and Cribb, A.E. (1999) Effects of Phenobarbital treatment on serum thyroxine and thyroid-stimulating hormone concentration in epileptic dogs. Journal of the American Veterinary Medical Association 215, 489–496.
Gaskill, C.L., Burton, S.A., Gelens, H.C., Ihle, S.L., Miller, J.B., Shaw, D.H., Brimacombe, M.B. and Cribb, A.E. (2000) Changes in serum thyroxine and thyroid-stimulating hormone concentrations in epileptic dogs receiving phenobarbital for one year. Journal of Veterinary Pharmacology and Therapeutics 23, 243–249.
Gaskill, C.L., Hoffman, W.E. and Cribb, A.E. (2004) Serum alkaline phosphatase isoenzyme profiles in phenobarbital-treated epileptic dogs. Veterinary Clinical Pathology 33, 215–222.
Gaskill, C.L., Miller, L.M., Mattoon, J.S., Hoffmann, W.E., Burton, S.A., Gelens, H.C., Ihle, S.L., Miller, J.B., Shaw, D.H. and Cribb, A.E. (2005) Liver histopathology and liver and serum alanine aminotransferase and alkaline phosphatase activities in epileptic dogs receiving phenobarbital. Veterinary Pathology 42, 147–160.
Gieger, T.L., Hosgood, G., Taboada, J., Wolfsheimer, K.J. and Mueller, P.B. (2000) Thyroid function and serum hepatic enzyme activity in dogs after phenobarbital administration. Journal of Veterinary Internal Medicine 14, 277–281.
Graham, R.A., Downey, A., Mudra, D., Krueger, L., Carroll, K., Chengelis, C., Madan, A. and Parkinson, A. (2002) In vivo and in vitro induction of cytochrome P450 enzymes in beagle dogs. Drug Metabolism and Disposition 30(11), 1206–1213.
Habock, G. and Pakozdy, A. (2012) Haematological abnormalities in dogs during Phenobarbital treatment. Wiener Tierärztliche Monatsschrift – Veterinary Medicine Austria 99, 18–25.
Hauptmann, A. (1912) Luminal bei Epilepsie. Munch Med Wochenschr 59, 1907–1909.
Hermann, R. and von Richter, O. (2012) Clinical evidence of herbal drugs as perpetrators of pharmacokinetic drug interactions. Planta Medica 78(13), 1458–1477.
Hess, R.S., Kass, P.H., Shofer, F.S., Van Winkle, T.J. and Washabau, R.J. (1999) Evaluation of risk factors for fatal acute pancreatitis in dogs. Journal of the American Veterinary Medical Association 214, 46–51.
Hojo, T., Ohno, R., Shimodo, M. and Kokue, E. (2002) Enzyme and plasma protein induction by multiple oral administrations of phenobarbital at a therapeutic dosage regimen in dogs. Journal of Veterinary Pharmacology and Therapeutics 25, 121–127.
Huang, Z. (2012) Effect of Alpha-1-Acid Glycoprotein Binding on Pharmacokinetics and Pharmacodynamics. Current Drug Metabolism Oct 15. [Epub ahead of print]
Jacobs, G., Calvert, C. and Kaufman, A. (1998) Neutropenia and thrombocytopenia in three dogs treated with anticonvulsivants. Journal of the American Veterinary Medical Association 212, 681–684.
Jamali, F. and Axelson, J.E. (1978) Griseofulvin–phenobarbital interaction: a formulation-dependent phenomenon. Journal of Pharmaceutical Sciences 67, 466–470.
Kantrowitz, L.B., Peterson, M.E., Trepanier, L.A., Melián, C. and Nichols, R. (1999) Serum total thyroxine, total triiodothyronine, free thyroxine, and thyrotropin concentrations in epileptic dogs treated with anticonvulsants. Journal of the American Veterinary Medical Association 214, 1804–1808.
Karalis, V., Macheras, P. and Bialer, M. (2013) Generic Products of Antiepileptic Drugs: A Perspective on Bioequivalence, Bioavailability, and Formulation Switches Using Monte Carlo Simulations. CNS Drugs. Oct 3. [Epub ahead of print]
Kennerly, E.M., Idaghdour, Y., Olby, N.J., Munana, K.R. and Gibson, G. (2009) Pharmacogenetic association study of 30 genes with phenobarbital drug response in epileptic dogs. Pharmacogenetics and Genomics 19, 911–922.
Khoutorsky, A. and Bruchim, Y. (2008) Transient leucopenia, thrombocytopenia, and anaemia associated with severe acute Phenobarbital intoxication in a dog. Journal of Small Animal Practice 49, 367–369.
Kluger, E.K., Malik, R., Ilkin, W.J., Snow, D., Sullivan, D.R. and Govendir, M. (2008) Serum triglyceride concentration in dogs with epilepsy treated with Phenobarbital or with Phenobarbital and bromide. Journal of the American Veterinary Medical Association 233, 1270–1277.
Kube, S.A., Vernau, K.M. and LeCouteur, R.A. (2006) Dyskinesia associated with oral phenobarbital administration in a dog. Journal of Veterinary Internal Medicine 20, 1238–1240.
Levitski, R.E. and Trepanier, L.A. (2000) Effect of timing of blood collection on serum phenobarbital concentration in dogs with epilepsy. Journal of the American Veterinary Medical Association 217, 200–204.
Lord, L.K. and Podell, M. (1999) Owner perception of the care of long-term phenobarbital-treated epileptic dogs. Journal of Small Animal Practice 40(1), 11–15.
Maguire, P.J., Fettman, M.J., Smith, M.O., Greco, D.S., Turner, A.S., Walton, J.A. and Ogilvie, G.K. (2000) Effects of diet on pharmacokinetics of phenobarbital in healthy dogs. Journal of the American Veterinary Medical Association 217, 847–852.
March, P.A., Hillier, A., Weisbrode, S.E., Mattoon, J.S., Johnson, S.E., DiBartola, S.P. and Brofman, P.J. (2004) Superficial necrolytic dermatitis in 11 dogs with a history of phenobarbital administration (1995–2002). Journal of Veterinary Internal Medicine 18, 65–74.
Mealey, K.L. (2004) Therapeutic implications of the MDR-1 gene. Journal of Veterinary Pharmacology and Therapeutics 27, 257–264.
Mealey, K.L., Bentjen, S.A., Gay, J.M. and Cantor, G.H. (2001) Ivermectin sensitivity in Collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics 11, 727–733.
Mizukami, K., Yabuki, A., Chang, H.S., Uddin, M.M., Rahman, M.M., Kushida, K., Kohyama, M. and Yamato, O. (2013) High Frequency of a Single Nucleotide Substitution (c.-6-180T>G) of the Canine MDR1/ABCB1 Gene Associated with Phenobarbital-Resistant Idiopathic Epilepsy in Border Collie Dogs. Disease Markers 35(6), 669–672.
Monteiro, R., Anderson, T.J., Innocent, G., Evans, N.P. and Penderis, J. (2009) Variations in serum concentration of PB in dogs receiving regular twice daily doses in relation to the times of administration. The Veterinary Record 165, 556–558.
Moore, S.A., Muñana, K.R., Papich, M.G. and Nettifee-Osborne, J.A. (2011) The pharmacokinetics of levetiracetam in healthy dogs concurrently receiving phenobarbital. Journal of Veterinary Pharmacology and Therapeutics 34, 31–34.
Morton, D.J. and Honhold, N. (1988) Effectiveness of a therapeutic drug monitoring service as an aid to the control of canine seizures. The Veterinary Record 122, 346–349.
Müller, P.B., Wolfsheimer, K.J., Taboada, J., Hosgood, G., Partington, B.P. and Gaschen, F.P. (2000a) Effects of long-term phenobarbital treatment on the thyroid and adrenal axis and adrenal function tests in dogs. Journal of Veterinary Internal Medicine 14, 157–164.
Müller, P.B., Taboada, J., Hosgood, G., Partington, B.P., VanSteenhouse, J.L., Taylor, H.W. and Wolfsheimer, K.J. (2000b) Effects of long term phenobarbital treatment on the liver in dogs. Journal of Veterinary Internal Medicine 14, 165–171.
Muñana, K.R., Nettifee-Osborne, J.A., Bergman, R.L. Jr and Mealey, K.L. (2012) Association between ABCB1 genotype and seizure outcome in collies with epilepsy. Journal of Veterinary Internal Medicine 26, 1358–1364.
Nardou, R., Yamamoto, S., Bhar, A., Burnashev, N., Ben-Ari, Y. and Khalilov, I. (2011) Phenobarbital but not Diazepam reduces AMPA/kainate receptor mediated currents and exerts opposite actions on initial seizures in the neonatal rat hippocampus. Frontier in Cellular Neuroscience 5, 16.
Orito, K., Saito, M., Fukunaga, K., Matsuo, E., Takikawa, S., Muto, M., Mishima, K., Egashira, N. and Fujiwara, M. (2008) Pharmacokinetics of zonisamide and drug interaction with phenobarbital in dogs. Journal of Veterinary Pharmacology and Therapeutics 31, 259–264.
Outerbridge, C.A., Marks, S.L. and Rogers, Q.R. (2002) Plasma amino acid concentrations in 36 dogs with histologically confirmed superficial necrolytic dermatitis. Veterinary Dermatology 13, 177–186.
Pakozdy, A., Sarchahi, A.A., Leschnik, M., Tichy, A.G., Halasz, P. and Thalhammer, J.G. (2013) Treatment and long-term follow-up of cats with suspected primary epilepsy. Journal of Feline Medicine and Surgery 15(4), 267–273.
Pedersoli, W.M., Wike, J.S. and Ravis, W.R. (1987) Pharmacokinetics of single doses of phenobarbital given intravenously and orally to dogs. American Journal of Veterinary Research 48, 679–683.
Podell, M. (2004) Seizures. In: Platt, S.R. and Olby, N.J. (eds) BSAVA Manual of Canine and Feline Neurology, 3rd edn. British Small Animal Veterinary Association, Quedgeley, Gloucester, UK, pp. 105–106.
Podell, M. and Fenner, W.R. (1993) Bromide therapy in refractory canine idiopathic epilepsy. Journal of Veterinary Internal Medicine 7, 318–327.
Podell, M. and Hadjiconstantinou, M. (1999) Low concentrations of cerebrospinal fluid GABA correlate to a reduced response to phenobarbital therapy in primary canine epilepsy. Journal of Veterinary Internal Medicine 13, 89–94.
Quesnel, A.D., Parent, J.M., McDonell, W., Percy, D. and Lumsden, J.H. (1997) Diagnostic evaluation of cats with seizure disorders: 30 cases (1991-1993). Journal of the American Veterinary Medical Association 210, 65–71.
Ravis, W.R., Pedersoli, W.M. and Wike, J.S. (1989) Pharmacokinetics of phenobarbital in dogs given multiple doses. American Journal of Veterinary Research 50, 1343–1347.
Rusbridge, C. (2013) Choosing the right drug. 1. Anticonvulsants used for first-line therapy. In Practice 35, 106–113.
Schwartz-Porsche, D. and Kaiser, E. (1989) Feline epilepsy. Problems in Veterinary Medicine 1, 628–649.
Schwartz-Porsche, D., Loscher, W. and Frey, H.H. (1985) Therapeutic efficacy of phenobarbital and primidone in canine epilepsy: a comparison. Journal of Veterinary Pharmacology and Therapy 8, 113–119.
Shah, S.S., Sanda, S., Regmi, N.L., Sasaki, K. and Shimoda, M. (2007) Characterization of cytochrome P450mediated drug metabolism in cats. Journal of Veterinary Pharmacology and Therapeutics 30(5), 422–428.
Shaik, I.H. and Mehvar, R. (2010) Cytochrome P450 induction by phenobarbital exacerbates warm hepatic ischemia reperfusion injury in rat livers. Free Radical Research 44, 441–453.
Shihab, N., Bowen, J. and Volk, H.A. (2011) Behavioral changes in dogs associated with the development of idiopathic epilepsy. Epilepsy and Behaviour 21(2), 160–167.
Solomon, G.E., Hilgartner, M.W. and Kutt, H. (1974) Phenobarbital-induced coagulation defects in cats. Neurology 24, 920–924.
Sridar, C., Goosen, T.C., Kent, U.M., Williams, J.A. and Hollenberg, P.F. (2004) Silybin inactivates cytochromes P450 3A4 and 2C9 and inhibits major hepatic glucuronosyltransferases. Drug Metabolism and Disposition 32(6), 587–594.
Steiner, J.M., Xenoulis, P., Anderson, J.A., Barr, A.C. and Williams, D.A. (2008) Serum pancreatic lipase immunoreactivity concentration in dogs treated with potassium bromide and/or Phenobarbital. Veterinary Therapeutics 9, 37–44.
Thompson, J.C. and Johnstone, A.C. (1983) Myelofibrosis in the dog: three case reports. Journal of Small Animal Practice 24, 589–601.
Thurman, G.D., McFayden, M.L. and Miller, R. (1990) The pharmacokinetics of phenobarbitone in fasting and non-fasting dogs. Journal of the South Africa Veterinary Association 61, 86–89.
Ticku, M.K. and Kulkarni, S.K. (1992) Antiepileptic actions of barbiturates. In: Faingold, C.L. and Fromm, G.H. (eds) Drugs for the Control of Epilepsy: Actions on Neuronal Networks Involved in Seizure Disorders. Boca Raton, Florida.
Trepanier, L.A., Van Schoick, A., Schwark, W.S. and Carrillo, J. (1998) Therapeutic serum drug concentrations in epileptic dogs treated with potassium bromide alone or in combination with other anticonvulsants: 122 cases (1992-1996). Journal of the American Veterinary Medical Association 213, 1449–1453.
Volk, H., Coleing, J.A.L., Platt, S.R. and Chandler, K. (2007) Clinical presentation and response to treatment of cats with epilepsy. In: Proceedings of British Small Animal Veterinary Association Congress, pp. 487–488.
Von Klopmann, T., Boettcher, I.C., Rotermund, A., Rohn, K. and Tipold, A. (2006) Euthyroid sick syndrome in dogs with idiopathic epilepsy before treatment with anticonvulsant drugs. Journal of Veterinary Internal Medicine 20, 516–522.
Wagner, S.O., Sams, R.A. and Podell, M. (1998) Chronic phenobarbital therapy reduces plasma benzodiazepine concentrations after intravenous and rectal administration of diazepam in the dog. Journal of Veterinary Pharmacology and Therapeutics 21, 335–341.
Webster, C.R.L. and Cooper, J. (2009a) Diagnostic approach to hepatobiliary disease. In: Bonagura, J.D. and Twedt, D.C. (eds) Kirk’s Current Veterinary Therapy XIV. Saunders Elsevier, Missouri, pp. 543–549. Webster, C.R.L. and Cooper, J. (2009b) Therapeutic Use of Cytoprotective Agents in Canine and Feline Hepatobiliary Disease. Veterinary Clinics of North America - Small Animal Practice 39, 631–652.
Weiss, D. (2005) Bone marrow necrosis in dogs: 34 cases (1996-2004). Journal of the American Veterinary Medical Association 227, 263–267.
Weiss, D.J. and Smith, S.A. (2002) A retrospective study of 19 cases of canine myelofibrosis. Journal of Veterinary Internal Medicine 16, 174–178.
14 Bromide
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Bromide (Br) is a halide salt first used in the treatment of human epilepsy in 1857 (Locock, 1857). Although it is still used to treat specific types of refractory seizures in children, the use of Br in humans has decreased throughout the 20th century due to the availability of other anti-epileptic medications (AEMs) with fewer adverse effects (Friedlander, 2000; Korinthenberg et al., 2007; Djuric et al., 2011; Ünver et al., 2013).
The use of potassium bromide (KBr) was first reported in dogs in 1907 (Swell, 1907). Br can be administered as a potassium (K) or sodium (Na) salt, with 1 g of KBr containing less Br (67% Br, 670 mg) than 1 g of NaBr (78% Br, 780 mg) (Boothe, 2012). KBr is the most commonly used form. The use of NaBr (instead of KBr) has been recommended in dogs with renal dysfunction or adrenal insufficiency and when intravenous administration is required (Trepanier, 1995). NaBr is contraindicated in dogs with congestive heart failure, hypertension and hepatic failure (Trepanier, 1995).
KBr is an affordable, relatively safe and effective AEM for the long-term management of epilepsy in dogs. Its prolonged half-life allows for convenient once or twice daily administration. Br can be used as both a monotherapeutic agent or in combination with other AEMs. A recent double-blinded, randomized, parallel, clinical trial in idiopathic epileptic dogs did not support the preferential use of KBr over phenobarbital (PB) as the first choice AEM (Boothe, 2012 et al.). However, KBr may represent the first choice AEM in dogs with hepatic dysfunction or in dogs with concurrent disorders requiring life-long administration of other hepatically metabolized medications. As an adjunct to PB, KBr is efficacious in reducing seizure frequency in PB-resistant dogs and in maintaining seizure control in dogs requiring reduction of PB dosage due to intolerable adverse effects associated with its use (Podell and Fenner, 1993; Trepanier et al., 1998).
The use of Br is currently not recommended in epileptic cats due to the potential for life-threatening adverse effects and questionable efficacy (Wagner, 2001; Boothe et al., 2002; Bertolani et al., 2012).
KBr is licensed for veterinary use (as adjunctive AEM) in epileptic dogs in the UK and available as 325 mg tablets. In other countries, including the USA, there is no approved commercially available product and Br is compounded into a solution, capsules, or tablets from either the potassium-derived or sodium-derived analytic grade chemical.
Mechanism of Action
The exact mechanism by which Br exerts its antiseizure activity is incompletely understood.
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
The action seems to involve GABA receptor-gated chloride ion channels (Fig. 14.1). One proposed mechanism involves Br intracellular influx through the chloride ion channels with subsequent neuronal hyperpolarization, increase in seizure threshold and prevention of seizure discharge extension (Pearce, 1990; Podell and Fenner, 1993; Trepanier, 1995). Br-mediated enhancement of GABA-activated currents and potentiation of GABA-inhibitory postsynaptic potentials have been identified in vitro (Suzuki et al., 1994). Br may have a synergistic action with PB in raising seizure threshold by enhancing GABA-ergic activity.
Metabolism and Pharmacokinetics
Bromide salts (KBr and NaBr) are rapidly absorbed from the small intestine with peak absorption achieved 1.5 h after oral administration in dogs (Van Dyke and Hastings, 1931). The prandial state of the animal should not affect Br absorption after oral administration. The estimated oral bioavailability of Br is 46% (Trepanier and Babish, 1995a). Br is unbound to plasma proteins and can diffuse freely across cellular membranes. Br distributes into the extracellular space and equilibrates with chloride into most tissues of the body to maintain a constant halide concentration (Wallace and Brodie, 1939). Maximal concentration in the CSF (87% of the serum concentration) occurs about 2.5 h after a single intravenous administration of Br in dogs (Greenberg et al., 1943). In another study, administration of a single intravenous dose of Br resulted in CSF Br levels of 83% of plasma levels from 24 h to 13 days post-administration in dogs (Wallace, 1938). CSF Br concentration at steady-state was 77% of the serum Br concentration after multiple oral administrations (30 mg/kg KBr every 12 h) in dogs (March et al., 2002).
Pharmacokinetic parameters of Br after intravenous and oral administration in healthy dogs in different studies are presented in Table 14.1. Discrepancies in some pharmacokinetic parameters among studies are likely due to differences in dietary chloride content, route and method of Br administration, Br assay

| Chloride | Pharmacokinetic parameters | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| content of | |||||||||||||
| the diet on | Cl/F | ||||||||||||
| Br dose | a dry matter | Cl | (ml/kg/ | AUC | Cmax | Css | |||||||
| Reference | administered | basis (%) | F (%) | Vd (l/kg) | (ml/kg/day) | day) | (mg/l/day) | (µg/ml) | Tmax (h) | T½ (d) | (mg/dl) | ||
| Palmer and | 466 mg/kg IV | NA | Mean | NA | NA | NA | NA | NA | NA | NA | 33 | NA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clarke, | once | ||||||||||||||||||||
| 1933 | 390 mg/kg PO | NA | Mean | NA | NA | NA | NA | NA | NA | NA | 39 | NA | |||||||||
| once | |||||||||||||||||||||
| Schwartz | 20-27 mg/kg | NA | Mean | NA | NA | NA | NA | NA | NA | NA | 25 | NA | |||||||||
| Porsche | PO for | ||||||||||||||||||||
| et al., 1990 | 62 days | ||||||||||||||||||||
| Trepanier and | 20 mg/kg IV | 0.4 | Mean ± SD | NA | 0.45 ± 0.07 | 9 ± 3.9 | NA | 2484 ± 813 | NA | NA | 37 ± 10 | NA | |||||||||
| Babish, | once | ||||||||||||||||||||
| 1995a | 20 mg/kg PO | 0.4 | Mean ± SD | 46 | NA | NA | NA | 1133 ± 482 | 79 ± 13 0.69 ± 0.13 | 46 ± 9 | NA | ||||||||||
| once | |||||||||||||||||||||
| Trepanier and | 14 mg/kg PO | 0.2 | Mean ± SD | NA | NA | NA | NA | 1948 ± 1143 80 ± 12 | 0.6 ± 0.4 | 69 ± 22 | NA | ||||||||||
| Babish, | once | ||||||||||||||||||||
| 1995b | 14 mg/kg PO | 0.4 | Mean ± SD | NA | NA | NA | NA | 1270 ± 356 88 ± 8 | 1.0 ± 0.7 | 46 ± 6 | NA | ||||||||||
| once | |||||||||||||||||||||
| 14 mg/kg PO | 1.3 | Mean ± SD | NA | NA | NA | NA | 309 ± 147 80 ± 15 | 0.7 ± 0.4 | 24 ± 7 | NA | |||||||||||
| once | |||||||||||||||||||||
| Podell et al., | 1200 mg/kg/day | NA | NA | NA | 0.39 | 12.44 | NA | NA | 3193 | NA | 20.8 | NA | |||||||||
| 2000 | IV CRI | ||||||||||||||||||||
| 3%NaBr | |||||||||||||||||||||
| 1500 mg/kg/day | NA | NA | NA | 0.42 | 14.86 | NA | NA | 3524 | NA | 19.5 | NA | ||||||||||
| IV CRI | |||||||||||||||||||||
| 3%NaBr | |||||||||||||||||||||
| March et al., | 20 mg/kg q12h | 0.55 – 0.72 | Median | NA | 0.4 | 8.2 | 16.4 | NA | NA | NA | 15.2 | 245 | |||||||||
| 2002 | PO for 115 days | Range | NA | 0.32 – 0.46 | 6.0 – 12.6 | 14.9 – 22.5 | NA | NA | NA | 12.2 – 20.3 178 – 269 | |||||||||||
Br, bromide; PO, per os; IV, intravenously; F, bioavailability; Vd, volume of distribution; Cl, clearance; Cl/F, total body clearance; Vd/F, volume of distribution/extent of absorption; AUC, area under the concentration time curve; C, serum maximum concentration; T, time to maximum serum concentration; T½, elimination half-life; Css, steady-state serum concentration; NA, not available
maxmax
methodology as well as inter-individual variability in dogs (March et al., 2002).
Due to the prolonged elimination half-life of Br, steady state is reached approximately 2.5–3 months after treatment initiation at maintenance dosage (Podell and Fenner, 1993; Trepanier and Babish, 1995a; Podell, 1998; Ducoté, 1999; March et al., 2002). However, in one study, the majority of dogs administered KBr at 30 mg/kg every 12 h (60 mg/kg/ day, which is a higher dosage than routinely recommended for clinical use) reached 75% and 90% of apparent steady-state concentrations by 30 and 60 days, respectively (March et al., 2002).
Br crosses the placenta and is excreted in milk (Vobecký et al., 2005). Br is not metabolized and is excreted unchanged in the urine predominantly by glomerular filtration (Palmer and Clarke, 1933). Small amounts of Br are also excreted as saliva, sweat, nasal and conjunctival secretions and faeces containing unabsorbed gastric secretions (Bodansky and Modell, 1941). After glomerular filtration, Br ions are extensively reabsorbed by the renal tubules in competition with chloride ions (Wolf and Eadie, 1950). Consequently the rate of elimination and half-life of Br vary proportionally and inversely to chloride intake (Czerwinski, 1958; Trepanier and Babish, 1995b; Shaw et al., 1996). In a study evaluating the effect of dietary chloride on Br elimination in dogs, increasing dietary chloride from 0.2 to 1.3% on a dry matter basis resulted in a significant decrease in the mean apparent elimination half-life from 69 ± 22 to 24 ± 7 days (Trepanier and Babish, 1995b). In addition, the mean area under the concentration curve (AUC) for dogs fed 1.3% chloride was significantly smaller than the mean AUC of dogs fed 0.2% chloride. C and T were not sig
maxmax
nificantly different in dogs with different dietary chloride levels (Trepanier and Babish, 1995b). Predicted steady-state serum Br concentrations were significantly lower in dogs fed 1.3% chloride than in dogs fed 0.2% chloride (Trepanier and Babish, 1995b). The predicted mean daily dose of Br needed to maintain serum levels above 1 mg/ml for dogs fed 1.3% chloride (43 ± 13 mg/kg) was nearly twice as high as the dose estimated for dogs fed 0.4% chloride (22 ± 3 mg/kg) and approximately three times higher than the dose estimated for dogs fed 0.2% chloride (15 ± 4 mg/kg). These differences were statistically significant (Trepanier and Babish, 1995b).
The pharmacokinetics of Br in dogs administered a loading dose of 600 mg/kg of sterile KBr solution (concentration 250 mg/kg) either intrarectally (IR) as six boluses of 100 mg/kg every 4 h for 24 h or as constant rate infusion over 24 h have been investigated (Dewey et al., 1999; Boothe, 2012). The average peak serum Br concentration after IR loading was similar (0.91 mg/ml, range 0.81–1.11 mg/ ml) to the one reached after IV loading (1.10 mg/ml, range 0.89–1.22 mg/ml). The mean half-life of IR-administered Br was 20.4 days (Boothe, 2012). Bioavailability of IR Br was calculated as 57.7% in two dogs (Dewey et al., 1999) and as 107% in five dogs (Boothe, 2012).
In another study, a loading dose of 1200 or 1500 mg/kg/day at a continuous rate IV infusion of 3% NaBr resulted in peak serum Br concentration of 319.3 and 352.4 mg/dl, CSF serum ratio at 24 h of 0.65 and 0.68, clearance of 12.44 and 14.86 ml/day/kg, volume of distribution of 0.39 and 0.42 l/kg and elimination half-life of 20.8 and 19.5 days, respectively (Podell et al., 2000).
In cats administered 15 mg/kg of KBr orally every 12 h, the maximum serum Br concentration was 1.1 ± 0.2 mg/ml at 8 weeks, mean total body clearance was 0.21 ± 0.03 l/kg/week, mean elimination half-life of Br was approximately 12 days and steady state was achieved at a mean of 5.3 ± 1.1 weeks (Boothe et al., 2002).
Pharmacokinetic Interactions and Adverse Reactions
Pharmacokinetic interactions of Br are limited as it is not metabolized or protein-bound. The main pharmacokinetic interactions are associated with alterations in the renal excretion of Br. As mentioned above, the rate of elimination of bromide varies proportionally and inversely to chloride intake (Czerwinski, 1958; Trepanier and Babish, 1995b; Shaw et al., 1996).
An increase in chloride intake (e.g. diet change, ingestion of salty treats or sea water, administration of intravenous fluids or drug formulations containing chloride) will enhance renal elimination rate of Br and subsequently decrease serum Br concentration, potentially causing loss of seizure control (including occurrence of cluster seizures or status epilepticus) (Shaw et al., 1996). Loop diuretics (such as furosemide) may enhance Br elimination by blocking Br reabsorption through renal tubular chloride channels (Millns and Rogers, 1978). On the contrary, osmotic diuretics do not seem to affect Br excretion (Palmer and Clarke, 1933). A decrease in chloride intake (e.g. diet change) will result in reduced renal elimination rate of Br, subsequent increase in serum Br concentration and potentially cause bromide toxicity (bromism) (Rossmeisl and Inzana, 2009). Dogs administered KBr should be maintained on a constant diet (and overall chloride intake) to prevent fluctuations in serum bromide concentrations, which could result in therapeutic failure or toxicity.
KBr should be avoided in dogs with renal dysfunction to prevent toxicity secondary to reduced renal elimination (Nichols et al., 1996). If there are no alternatives to KBr, dogs with renal dysfunction should initially be administered half the recommended Br dosage with close monitoring for adverse effects along with frequent measurement of Br serum concentrations (Trepanier, 1995).
Small increases in serum Br concentration have been reported in dogs and humans following halothane anaesthesia (Pedersoli, 1980). Br is released from halothane during its metabolism by hepatic CYP450 and this production is enhanced by concurrent administration of PB. Although a single anaesthesia with halo-thane may not result in a clinically significant increase in serum Br concentration, it would be safer to use other inhalant anaesthetics in dogs on Br antiseizure treatment.
Transient and dose-related adverse effects of Br in dogs
Common adverse effects of Br in dogs include (Pearce, 1990; Podell and Fenner, 1993, 1994; Trepanier and Babisch, 1995a; Chang et al., 2006; Baird-Heinz et al., 2012):
Gastrointestinal irritation can be prevented or minimized by administering Br salts with food (Baird-Heinz et al., 2012) and dividing the daily dose into two or more doses. Solutions may be better tolerated than capsules in some individuals and NaBr may be better tolerated than KBr.
Bromide toxicity (bromism)
Bromide toxicity (bromism) is a clinically heterogeneous, dose-dependent neurotoxicosis that is reversible with treatment in the majority of dogs (Rossmeisl and Inzana, 2009). In one study, the prevalence of bromism in dogs with idiopathic epilepsy was 2% and the development of bromism was significantly associated with total daily bromide dose (mean 44.9 ± 1.7 mg/kg) and serum Br concentration (mean 3.7 ± 0.3 mg/ml) at admission, suboptimal monitoring of serum Br concentrations (mean interval 556 ± 54 days) and dietary chloride content (Rossmeisl and Inzana, 2009). Br toxicosis can result from diminished Br excretion due to development of renal insufficiency, a chloride-deficient diet, increased Br intake due to misformulation of Br solution or capsules, accidental overdosing, ingestion of spa water, well water or medications containing Br (Trepanier, 1995; Nichols et al., 1996; Rossmeisl and Inzana, 2009). There is individual variability in the degree of clinical tolerance to Br and therefore different dosages and serum levels can be associated with toxicosis (Rosemblum, 1958; Yohn et al., 1992; March et al., 2002). Most commonly, bromism is associated with serum Br concentrations near or above the upper end of the reference range (March et al., 2002; Rossmeisl and Inzana, 2009). Clinical signs of bromism are generally subacute to chronic progressive and include obtundation, stupor or coma, bilateral mydriasis with slow and incomplete pupillary light reflexes, bilateral blindness, anisocoria, abnormal behaviour, head pressing, ataxia, paraparesis, tetraparesis with normal or decreased spinal reflexes, dysphagia, megaoesphagus and muscle pain (Yohn et al., 1992; Rossmeisl and Inzana, 2009).
Clinical, electrodiagnostic and histologic findings of Br-associated neuromyopathy have been reported (Steinmetz et al., 2012). Mild cases of bromism can be treated with dose reduction, generally by 25% to 50%, and a clinical response should be seen within 1 to 2 weeks. More severe cases are managed by temporarily stopping the Br and diuresis with intravenous saline (0.9% NaCl) to promote Br excretion and gradually reduce serum concentrations. It has been reported that IV administration of 0.9% NaCl at a maintenance rate for <24 h is typically sufficient to resolve clinical signs of bromism (Trepanier, 1995). Furosemide has also been used to promote Br diuresis, but its impact on Br clearance is unclear (Boothe, 2012). Sudden and excessive reduction in serum Br concentrations can lead to breakthrough seizures, therefore the diuresis should be done carefully in conjunction with serial monitoring of serum Br concentrations. A lower oral dose of bromide can be started once the signs of toxicity resolve. If bromism is related to development of renal insufficiency, alternative AEM such as levetiracetam, zonisamide or phenobarbitone (see Chapters 13, 15, 16) should be considered and the KBr dosage should be reduced by 50% or discontinued.
Suspected idiosyncratic adverse effects of Br in dogs
Uncommonly reported adverse effects of Br in dogs include (Pearce, 1990; Podell and Fenner, 1993; Dowling, 1994; Trepanier, 1995; Gaskill and Cribb, 2000; March et al., 2002; Chang et al., 2006; Dewey, 2006; Baird-Heinz et al., 2012):
Personality changes
Behavioural changes such as fear/anxiety, defensive aggression and abnormal perception (e.g. barking without apparent cause, chasing shadows or light spots, aimless pacing, staring into space) can occur in dogs at the time of development of idiopathic epilepsy (Shihab et al., 2011). Therefore it may be difficult to differentiate neurobehavioural comorbidities of epilepsy from PB (or other AEM)-related behavioural abnormalities. Intolerable personality changes presumed to be caused by Br treatment generally subside or resolve following dose reduction or treatment discontinuation.
Pruritic dermatologic lesions
Pruritic dermatologic lesions have rarely been reported in dogs treated with Br and generally resolve following discontinuation of Br. Corticosteroid administration can rapidly resolve the pruritus. Bromoderma (pigmentation, erythematous dermatitis and nodular/pustular skin lesions) is also a rare adverse effect of Br in humans (Anzai et al., 2003).
Panniculitis
Panniculitis accompanied by lethargy and pyrexia has been reported in association with administration of KBr in two dogs (Boynosky and Stokking, 2013). Both dogs were treated with KBr for idiopathic epilepsy for over 1 year. Dose increases in both cases were associated with panniculitis, characterized by painful subcutaneous nodules in a generalized distribution over the trunk. Nodule eruption was persistent in one dog and waxed and waned in the other. In both cases, panniculitis, lethargy and pyrexia resolved within 7 days after discontinuation of KBr and did not recur in the following 2 years. It has been proposed that the rapid resolution of the panniculitis was due to a decrease in serum Br concentrations below a threshold that may be required to trigger the development of panniculitis. No other cause of panniculitis could be determined for either dog. Panniculitis has been reported after administration of KBr in people and may be a form of drug-induced erythema nodosum (Boynosky and Stokking, 2013).
Persistent cough
A persistent cough, possibly associated with a bronchial asthma-like condition, has been reported in some epileptic dogs treated with Br (Dewey, 2006). Diagnostic tests did not identify a cause for the cough. Coughing resolved shortly after Br discontinuation (Dewey, 2006).
Pancreatitis
Pancreatitis has been reported as a possible adverse effect in epileptic dogs treated with Br monotherapy or in combination with PB (Schwartz-Porsche and Jürgens, 1991; Podell and Fenner, 1993; Hess et al., 1999; Gaskill and Cribb, 2000). In one study, estimated prevalence of pancreatitis was at least 10% in dogs treated with PB and KBr (Gaskill and Cribb, 2000). However, a cause and effect relationship has not been proven yet. It has been speculated that polyphagia secondary to PB and/or KBr could lead to dietary indiscretion and consequently to pancreatitis (Podell and Fenner, 1993). An increased risk for elevated serum canine pancreatic lipase immunoreactivity concentrations has been reported in epileptic dogs treated with Br or PB alone or in combination (Steiner et al., 2008). Fasting hypertriglyceridaemia has been associated with high postprandial serum triglyceride concentrations, which is a known risk factor for pancreatitis. Fasting hypertriglyceridaemia was identified in 41% (12/29) of epileptic dogs treated with PB and KBr (Kluger et al., 2008); 17% (5/29) of these dogs had a history of pancreatitis. Serum canine pancreatic lipase immunoreactivity was measured in 16 of the 29 dogs treated with PB and KBr and was greater than the upper reference limit in six dogs (38%). Of these dogs, 72% (21/29) treated with PB and KBr had a body condition score ³6 (Kluger et al., 2008). To minimize the risk of pancreatitis in epileptic dogs treated with PB and KBr, it has been recommended to feed a low-fat diet, provide regular exercise to maintain a healthy body-condition score and to perform periodical monitoring of fasting serum triglyceride concentration (Kluger et al., 2008).
Br-associated lower airway disease in cats
Br-associated lower airway disease has been reported in cats by different investigators (Wagner, 2001; Boothe et al., 2002; Volk et al., 2006; Bertolani et al., 2012). The prevalence has ranged from 35% to 67% of cats administered Br (Boothe et al., 2002; Volk et al., 2006). Br treatment duration prior to onset of clinical signs has ranged from 2 weeks to 96 months. Clinical signs can include coughing, dyspnoea, tachypnoea, and increased respiratory sounds and wheezing on thoracic auscultation. Haematology may reveal mild eosinophilia. Thoracic radiographs demonstrate bronchial patterns sometimes with peribronchial cuffing. Cytological examination of bronchoalveolar lavage specimens reveals eosinophilic inflammation alone or in combination with neutrophilic inflammation. Histology revealed endogenous lipid pneumonia associated with suppurative bronchitis in one cat with nodular pulmonary lesions and pneumothorax considered secondary to Br-associated lower airway disease (Bertolani et al., 2012). No associations between occurrence of respiratory signs and Br dose, serum level or treatment duration have been identified (Boothe et al., 2002). This suggests an allergic reaction or hypersensitivity pathophysiology rather than a dose-dependent reaction to Br (Boothe et al., 2002). Respiratory signs usually improve with steroid treatment and resolve 1–2 months following discontinuation of KBr, however they may be life threatening and sometimes lead to euthanasia. Oxygen therapy and bronchodilators may be required in severely affected cats. Mortality as a result of Br-associated lower airway disease can be 14% to 18% of affected cats (Wagner, 2001; Boothe et al., 2002; Bertolani et al., 2012).
Br-related laboratory changes
Factitious hyperchloraemia
(pseudohyperchloraemia)
Administration of Br can result in factitious hyperchloraemia (pseudohyperchloraemia) when assaying serum, whole blood or plasma. Most commercially available biochemical analytical methods used for chloride determination actually measure the total halide ion concentration in the sample, which typically includes chloride, physiologic concentrations of endogenous Br and the exogenously administered Br (Woody et al., 1990). Unlike true hyperchloraemia, pseudohyperchloraemia is not associated with hypernatraemia or metabolic acidosis (Trepanier, 1995).
Effect on thyroid function tests
Chronic KBr treatment does not result in any significant change in canine thyroid function or morphology (Kantrowitz et al., 1999; Paull et al., 2003).
Dosing and Monitoring Recommendations
Routine initiation
Initial oral dosages of KBr as maintenance therapy in dogs are:
Because of the long elimination half-life, KBr can be administered once daily; however, twice-daily dosing as well as administration with food can help to prevent gastrointestinal mucosa irritation. NaBr may be less irritating to the gastric mucosa and may be indicated in dogs that experience nausea or vomiting secondary to KBr-induced gastric mucosa irritation (Trepanier, 1995).
If NaBr is used, the dose should be approximately 15% less than that calculated for KBr as NaBr contains more Br per gram than KBr (Trepanier, 1995).
As with PB, unless seizure frequency is high, initiation of therapy should begin at the low end of the dose range. Decision on dosage modulation should be based on seizure control, presence of adverse effects and serum concentration monitoring.
Effect of diet
The dosages indicated above are adequate in most dogs fed diets containing approximately 0.5–1% chloride on a dry matter basis. A lower or higher Br dosage would be required in dogs fed diets with lower or higher chloride content, respectively. The predicted mean daily dose of KBr needed to maintain serum Br levels above 1 mg/ml for dogs fed diets containing 1.3, 0.4 and 0.2% chloride on dry matter was 65, 32 and 23 mg/kg/day, respectively (Trepanier and Babish, 1995b). Most canine commercial diets have a similar chloride content on a dry matter basis (0.5–1%; Fig. 14.2, Table 14.2) with the exception of prescription diets for heart disease or some hypoallergenic or intestinal diets, which have a lower content, and some diets for urinary stones or rehydration diets, which have a higher content. Home-prepared diets have variable salt content. Pet food’s chloride content is normally not listed on the food label. Therefore, the pet owner or the veterinarian needs to contact the dog food manufacturer to obtain this information as it may influence Br daily dose. Dog owners should be instructed that any diet change during Br treatment should be done only if necessary and under veterinary supervision as the Br dosage may need to be adjusted accordingly. The diet change

Sensitivity (Purina) Digestive lamb (Purina) Adult light (Purina) EN – Dry (Purina) Hypoallergenic (Purina) Joint Mobility (Purina) Dermatologic management (Purina) Kidney Function (Purina) Obesity Management – wet (Royal Canin) Obesity Management – dry (Royal Canin) Satiety (Royal Canin) Weight control (Royal Canin) Hypoallergenic (Royal Canin) Mobility special (Royal Canin) Dental (Royal Canin) Convalescence (Royal Canin) Rehydration (Royal Canin) Diabetic – wet (Royal Canin) Intestinal – wet (Royal Canin) Digestive – wet (Royal Canin) Renal – wet (Royal Canin) Urinary S/O – wet (Royal Canin) Cardiac – wet (Royal Canin)
Fig. 14.2. Chloride content as percentage on a dry matter basis of different canine commercial diets.
should be gradual, at least over 1 week. Serum Br-level monitoring should be performed just before and after the diet change.
Bromide used concurrently with PB
When Br is added to PB to improve seizure control and there are no PB-induced life-threatening adverse effects, PB is continued at the current dosage at least until steady-state reference serum Br level has been reached. Subsequently, if seizure control is satisfactory but adverse effects (e.g. sedation, ataxia, pelvic limb weakness, polydipsia, polyuria or polyphagia) persist, PB dosage may be decreased by 10–25% every 4–6 weeks while the dog is closely monitored clinically. Alternatively the Br dose can be decreased by 10–25%.
Loading dose regimen
Br can be administered at a loading dose when steady-state reference serum levels have to be reached as rapidly as possible (e.g. dogs with frequent or severe seizures, or when PB must be rapidly discontinued because of severe adverse effects). Different protocols have been reported. Oral loading can be performed by administering KBr at 625 mg/kg divided in eight or more doses over 48 h, or 125 mg/kg/day divided in three to four daily administrations for 5 consecutive days. Loading is often associated with adverse effects (e.g. nausea, vomiting, diarrhoea, sedation, ataxia and pelvic limb weakness, polydipsia, polyuria and polyphagia) and hospitalization of the animal is recommended (Trepanier, 1995). A loading dose of 600 mg/kg KBr divided into multiple administrations over 17–48 h in combination with a maintenance dosage of
Table 14.2. Chloride content of Hill’s diets (courtesy of Iain Grant, Hill’s Pet Nutrition Ltd).
| Chloride | Chloride (mg) | ||
|---|---|---|---|
| Chloride percentage | percentage | per 100 kcal | |
| Diet | dry matter basis | as fed | MEa |
Prescription Diet Range Hill’s Prescription Diet™ a/d™ canned Hill’s Prescription Diet™ b/d™ dry Hill’s Prescription Diet™ c/d™ dry Hill’s Prescription Diet™ c/d™ canned Hill’s Prescription Diet™ d/d™ canned Venison
and Potato Hill’s Prescription Diet™ d/d™ canned Duck and Potato Hill’s Prescription Diet™ d/d™ canned Salmon
and Potato Hill’s Prescription Diet™ d/d™ dry Salmon and Rice Hill’s Prescription Diet™ d/d™ dry Duck and Rice Hill’s Prescription Diet™ d/d™ dry Egg and Rice Hill’s Prescription Diet™ h/d™ dry Hill’s Prescription Diet™ h/d™ canned Hill’s Prescription Diet™ i/d™ dry Hill’s Prescription Diet™ i/d™ canned Hill’s Prescription Diet™ j/d™ dry Hill’s Prescription Diet™ j/d™ canned Hill’s Prescription Diet™ k/d™ dry Hill’s Prescription Diet™ k/d™ canned Hill’s Prescription Diet™ l/d™ dry Hill’s Prescription Diet™ l/d™ canned Hill’s Prescription Diet™ n/d™ canned Hill’s Prescription Diet™ r/d™ dry Hill’s Prescription Diet™ r/d™ canned Hill’s Prescription Diet™ s/d™ canned Hill’s Prescription Diet™ Canine Metabolic
Advanced Weight Solution canned Hill’s Prescription Diet™ t/d™ dry Hill’s Prescription Diet™ u/d™ dry Hill’s Prescription Diet™ u/d™ canned Hill’s Prescription Diet™ w/d™ dry Hill’s Prescription Diet™ w/d™ canned Hill’s Prescription Diet™ z/d™ ultra dry Hill’s Prescription Diet™ z/d™ ultra canned Hill’s Prescription Diet™ z/d™ low allergen dry
Science Plan Range Hill’s Science Plan™ Puppy canned Hill’s Science Plan™ Puppy Healthy
Development™ Mini dry Hill’s Science Plan™ Puppy Healthy Development™ Medium dry Hill’s Science Plan™ Puppy Healthy Development™ Lamb and Rice dry Hill’s Science Plan™ Puppy Healthy Development™ Large Breed dry Hill’s Science Plan™ Canine Adult canned
0.71
0.71
0.49
0.65
0.66
0.77
0.57
0.87
0.90
0.61
0.30
0.32
1.05
1.00
0.65
0.56
0.75
0.44
0.83
1.05
0.56
0.83
0.49
2.37
0.74
0.34
0.60
0.50
0.52
0.76
0.78
0.36
0.52
0.76
0.80
0.80
0.76
0.95
0.52 0.17 148
0.65 180
0.46 115
0.19 143
0.16 147
0.19 198
0.14 143
0.80 215
0.83 224
0.56 151
0.28 71
0.09 69
0.97 270
0.26 235
0.60 165
0.18 133
0.69 175
0.12 95
0.77 190
0.32 230
0.16 119
0.76 243
0.12 164
0.69 521
0.19 221
0.31 88
0.55 138
0.14 106
0.48 159
0.19 213
0.68 195
0.09 95
0.48 132
0.23 185
0.74 188
0.74 188
0.70 182
0.88 242
0.13 107
Continued Continued
| Chloride | Chloride (mg) | ||
| Chloride percentage | percentage | per 100 kcal | |
| Diet | dry matter basis | as fed | MEa |
| Savoury Turkey canned | |||
| Hill’s Science Plan™ Canine Adult | 0.60 | 0.55 | 146 |
| Advanced Fitness™ Mini dry | |||
| Hill’s Science Plan™ Canine Adult Advanced | 0.66 | 0.61 | 162 |
| Fitness™ Medium dry Chicken | |||
| Hill’s Science Plan™ Canine Adult Advanced | 0.43 | 0.40 | 107 |
| Fitness™ Medium dry Tuna and Rice | |||
| Hill’s Science Plan™ Canine Adult Advanced | 0.57 | 0.53 | 141 |
| Fitness™ Lamb and Rice dry | |||
| Hill’s Science Plan™ Canine Adult Advanced | 0.68 | 0.63 | 165 |
| Fitness™ Large Breed dry | |||
| Hill’s Science Plan™ Canine Adult Advanced | 0.59 | 0.54 | 145 |
| Fitness™ Large Breed Lamb and Rice dry | |||
| Hill’s Science Plan™ Canine Adult Light canned | 0.62 | 0.16 | 184 |
| Hill’s Science Plan™ Canine Adult Light dry | 0.45 | 0.41 | 135 |
| Hill’s Science Plan™ Canine Adult Light | 0.70 | 0.64 | 209 |
| Large Breed dry | |||
| Hill’s Science Plan™ Canine Adult | 0.98 | 0.91 | 215 |
| Performance dry | |||
| Hill’s Science Plan™ Canine Adult Oral | 0.71 | 0.65 | 187 |
| Care dry | |||
| Hill’s Science Plan™ Canine Adult Sensitive | 1.02 | 0.94 | 247 |
| Skin dry | |||
| Hill’s Science Plan™ Canine Adult Sensitive | 1.00 | 0.92 | 250 |
| Stomach dry | |||
| Hill’s Science Plan™ Canine Adult Healthy | 0.40 | 0.37 | 101 |
| Mobility dry | |||
| Hill’s Science Plan™ Canine Mature | 0.53 | 0.13 | 138 |
| Adult 7+ canned | |||
| Hill’s Science Plan™ Canine Mature | 0.48 | 0.44 | 119 |
| Adult 7+ Active Longevity Mini dry | |||
| Hill’s Science Plan™ Canine Mature | 0.48 | 0.44 | 119 |
| Adult 7+ Active Longevity Medium dry | |||
| Hill’s Science Plan™ Canine Mature | 0.50 | 0.46 | 125 |
| Adult 7+ Active Longevity Large Breed dry | |||
| Hill’s Science Plan™ Canine Mature | 0.57 | 0.52 | 140 |
| Adult 7+ Active Longevity Lamb and Rice dry | |||
| Hill’s Science Plan™ Canine Mature | 0.50 | 0.49 | 158 |
| Adult 7+ Light dry | |||
| Vet Essentials Range | |||
| Hill’s Science Plan™ Vet Essential Puppy dry | 0.81 | 0.75 | 191 |
| Hill’s Science Plan™ Vet Essential Puppy | 0.88 | 0.81 | 224 |
| Large Breed dry | |||
| Hill’s Science Plan™ Vet Essential | 0.73 | 0.67 | 193 |
| Canine Adult Mini dry | |||
| Hill’s Science Plan™ Vet Essential | 0.73 | 0.67 | 193 |
| Canine Adult dry | |||
| Hill’s Science Plan™ Vet Essential | 0.79 | 0.73 | 210 |
| Canine Adult Large Breed dry | |||
| Hill’s Science Plan™ Vet Essential | 0.59 | 0.55 | 180 |
| Canine Mature Adult Mini dry |
Table 14.2. Continued.
| Chloride | Chloride (mg) | ||
|---|---|---|---|
| Chloride percentage | percentage | per 100 kcal | |
| Diet | dry matter basis | as fed | MEa |
| Hill’s Science Plan™ Vet Essential | 0.59 | 0.55 | 180 |
|---|---|---|---|
| Canine Mature Adult dry | |||
| Nature’s Best Range | |||
| Hill’s Nature’s Best™Puppy Mini/Medium dry | 1.28 | 1.18 | 314 |
| Hill’s Nature’s Best™Puppy Large/Giant dry | 0.80 | 0.74 | 209 |
| Hill’s Nature’s Best™ Adult Mini/Medium dry | 0.35 | 0.32 | 87 |
| Hill’s Nature’s Best™ Adult Large/Giant dry | 0.80 | 0.74 | 202 |
| Hill’s Nature’s Best™ Mature Adult 7+ Mini/Medium dry | 0.47 | 0.44 | 121 |
aME, metabolizable energy. The nutrient content of food is usually expressed on an ‘as fed’, ‘dry matter’ or a ‘kilocalorie’ basis. As fed is exactly as it describes: ‘the content of a specific nutrient in the food as it is presented and fed to the dog or cat’. The dry matter calculation eliminates the effect of any difference in moisture content when comparing two foods, e.g. a can (may be as high as 85% water) versus a dry product (may contain as little as 5% water). By subtracting the water content of the food as fed, the amount of nutrient can be recalculated based on the amount contained in the remaining dry matter (dry matter basis). The more accurate analysis is the calculation which determines the amount of a nutrient on an energy basis as this method also eliminates differences in the energy content when comparing two foods,
e.g. high- versus low-calorie formulations. This is important since an animal eats to satisfy its energy requirement and will therefore consume a greater amount of a low-energy formula than a high-energy formula. A suitable, balanced and complete diet will provide the correct intake of all nutrients when the animal has eaten enough of it to meet its energy requirements. When comparing two foods which contain the same percentage dry matter content of (e.g. chloride) but having very different caloric densities, the animal’s absolute intake of chloride would be greater when fed the low-energy formula since it would have eaten a larger amount of it than if it were fed the high-energy formula. The energy basis nutrient content of a food is usually expressed as milligrams of nutrient per 100 kcal of metabolizable energy.
33 mg/kg/day KBr resulted in median serum Br concentrations of 1.26 mg/ml (0.74–3.6 mg/ml) 1–24 h after completion of loading in 38 dogs. The majority of dogs (84.2%) had serum Br concentrations between 1 and 3 mg/ml (Gindiciosi et al., 2014).
When Br loading is needed in dogs that are unable to take oral medication (e.g. status epilepticus, severe sedation and/or severe postictal phase) Br can be administered IV or IR (Dewey et al., 1999; Podell, 2005; Boothe, 2012). A 3% NaBr solution in sterile water administered as a constant rate infusion (CRI) over 24 h at a dose of 900 mg/kg has been suggested for intravenous loading in dogs (Podell, 2005). Intravenous administration of KBr is not recommended due to the potential risk of cardiac arrhythmias resulting from rapid potassium infusion (Trepanier, 1995). A loading protocol of 600 mg/kg of sterile KBr solution (concentration 250 mg/ml) administered IR either as six boluses of 100 mg/kg every 4 h over 24 h or as constant rate infusion over 24h has been proposed (Dewey et al., 1999; Boothe, 2012). Reported adverse effects included transient diarrhoea for 24–48 h and sedation.
The loading dose can be followed by the maintenance dosage.
A ‘mini’-loading dose can be used in animals already on KBr maintenance dosage that require a rapid increase in serum Br levels due to poor seizure control. The maintenance dosage of KBr will need to be increased as well. The total supplemental KBr loading dose can be calculated based on the following formula and administered in divided doses over 2–5 days:
Total supplemental KBr loading dose in mg/kg = Vd/F × (desired serum Br concentration in mg/l − current serum Br concentration in mg/l)
where Vd/F = apparent volume of distribution (0.4 l/kg).
The new maintenance dosage can be calculated by adding to the current dose an additional dose calculated as:
Additional dose (mg/kg/day) to existing dose = (desired serum Br concentration in mg/l − current serum Br concentration in mg/l) × Cl/F
where Cl/F = clearance/bioavailability = 0.02
Reference serum Br concentrations
Reference serum Br concentrations are approximately:
To convert Br concentration from mg/l to µmol/l the conversion factor is 0.0125 and the formula is:
Br concentration in µmol/l = 0.0125 × Br concentration in mg/l
As for PB, there is individual variability in serum concentrations associated with seizure control or toxicity and reference ranges should be used as a guideline when treatment is initiated. Subsequently the oral dosage should be individualized based on clinical response and serum-level monitoring. The lowest oral dose and serum drug concentration associated with seizure eradication or >50% reduction and absence of intolerable adverse effects should be identified and used for individual patients.
Monitoring serum Br concentrations
Serum Br concentrations should be monitored:
0.5 mg/ml increment in Br serum level (Boothe, 2012);
• once the desired clinical response has been achieved in order to establish the individual therapeutic range;
Serum Br concentrations can also be evaluated 1 month after treatment initiation at maintenance dosage to estimate serum level at steady state, which should be at least twice as high. If the estimated steady-state serum Br level is lower than the reference range and seizure control is inadequate, Br oral dosage can be increased.
Timing of blood sampling for therapeutic monitoring
Timing of blood sample collection for Br quantification relative to oral administration is not critical due to the long elimination half-life of Br (Trepanier, 1995).
Br-induced pseudohyperchloraemia as predictor of serum Br concentration
The magnitude of Br-induced pseudohyperchloraemia has been used in epileptic people as indirect estimator of the Br concentration (Woody et al., 1990). In epileptic dogs the degree of pseudohyperchloraemia has been reported as inadequate predictor of serum Br concentration for routine clinical application and therefore decision on dose adjustment should be based on serum Br concentration (Rossmeisl et al., 2006). However, as chloride levels can be rapidly evaluated in-house in most veterinary clinics whereas serum Br levels have to be evaluated in a specialized laboratory, the measured chloride value can be used to guide adjustment of Br dosage in emergency clinical situations (Rossmeisl et al., 2006).
Efficacy
Br has been shown to be efficacious as a sole AEM in epileptic dogs but is not as efficacious as PB (Boothe et al., 2012). In a recent double-blinded, randomized, parallel, clinical trial in dogs, Br monotherapy resulted in a significant decrease in seizure number and severity and an increase in seizure interval at study end (6 months), compared with baseline. In addition, seizure duration decreased over time, although not significantly (Boothe et al., 2012). Seizure activity was eradicated in 52% (12/23) and decreased by >50% in 65% (15/23) of Br-treated dogs. Mean serum Br concentration was 1.8 ± 0.6 mg/ ml (range 0.9 to 3.3 mg/ml) in dogs with seizure eradication and 2.1 ± 0.6 mg/ml (range 1.5–2.7 mg/ml) in dogs with seizure reduction >50%. Number of seizures per month increased in 13% (3/23) of Br-treated dogs with serum Br concentrations of 1.8, 2.5 and 2.9 mg/ml, respectively. The correlation between Br dosage and either serum Br concentrations or treatment response was poor. PB was superior to Br in all outcome measures. In addition, the proportion of dogs with sedation and vomiting at study end was greater in the Br than PB treatment group (Boothe et al., 2012).
Br has been shown to be an effective add-on AEM in idiopathic epileptic dogs resistant to PB. The addition of KBr reduced seizure frequency in approximately 55% to 83% of dogs with approximately 21% to 26% attaining seizure-free status (Schwartz-Porsche and Jürgens, 1991; Podell and Fenner, 1993; Trepanier et al., 1998). Addition of KBr to PB also resulted in a decrease in seizure severity, intensity and tendency to occur in clusters (Pearce, 1990; Podell and Fenner, 1993). KBr has also been effective in maintaining seizure control in dogs requiring reduction or discontinuation of PB due to unacceptable adverse effects (Pearce, 1990; Trepanier, 1995; Trepanier et al., 1998). In one study, PB or primidone could eventually be discontinued in 19% of dogs administered KBr while improved seizure control was maintained (Trepanier et al., 1998). In addition, PB dose could be decreased by a mean of 47% (range 6–84%) in 34% of dogs with improved seizure control after serum Br reference serum concentrations were reached (Trepanier et al., 1998). The mean serum Br concentration was significantly higher (2.043 ±
0.792 mg/ml) in dogs in which PB could be discontinued than in dogs in which PB could only be decreased (1.657 ± 0.519 mg/ml) or was unchanged (1.470 ± 0.592 mg/ml) (Trepanier et al., 1998).
Dogs with inadequate seizure control despite serum levels of both PB and KBr or KBr alone (if PB is contraindicated) in the higher reference range for at least 2 months or with persistent intolerable adverse effects are considered resistant to these medications and require additional or alternative AEM (see Chapters 12, 15–20 and 22).
KBr seems to be less efficacious as an AEM in cats compared to dogs. One study reported that seizures were controlled only in 7 of 14 treated cats (Boothe et al., 2002). In another study, five of nine cats had no seizure during the trial (Volk et al., 2006). However, in these studies 35% to 67% of cats developed adverse effects and KBr had to be discontinued in several cats.
Summary Recommendations
to reach steady-state concentrations and therefore maximum clinical effect.
to prevent fluctuations in serum Br concentrations that can result in loss of seizure control or toxicity.
References
Anzai, S., Fujiwara, S. and Inuzuka, M. (2003) Bromoderma. International Journal of Dermatology 42, 370–371.
Baird-Heinz, H.E., Van Schoick, A.L., Pelsor, F.R., Ranivand, L. and Hungerford, L.L. (2012) A systematic review of the safety of potassium bromide in dogs. Journal of The American Veterinary Medical Association 15, 705–715.
Bertolani, C., Hernandez, J., Gomes, E., Cauzinille, L., Poujade, A. and Gabriel, A. (2012) Bromide-associated lower airway disease: a retrospective study of seven cats. Journal of Feline Medicine and Surgery 14, 591–597.
Bodansky, O. and Modell, W. (1941) The differential excretion of bromide and chloride ions and its role in bromide retention. Journal of Pharmacology and Experimental Therapeutics 73, 51–64.
Boothe, D.M. (1998) Anticonvulsant therapy in small animals. Veterinary Clinics of North America - Small Animal Practice 28, 411–444.
Boothe, D.M. (2012) Anticonvulsant and other neurologic therapies in small animals. In: Boothe, D.M. (ed.) Small Animal Clinical Pharmacology and Therapeutics, 2nd edn. Elsevier, St Louis, Missouri, pp. 950–955.
Boothe, D.M., George, K.L. and Couch, P. (2002) Disposition and clinical use of bromide in cats. Journal of the American Veterinary Medical Association 221, 1131–1135.
Boothe, D.M., Dewey, C. and Carpenter, D.M. (2012) Comparison of phenobarbital with bromide as a first-choice antiepileptic drug for treatment of epilepsy in dogs. Journal of the American Veterinary Medical Association 240, 1073–1083.
Boynosky, N.A. and Stokking, L.B. (2013) Potassium bromide-associated panniculitis. Journal of Small Animal Practice Sep 6. doi: 10.1111/jsap.12129. [Epub ahead of print]
Chang, Y., Mellor, D.J. and Anderson, T.J. (2006) Idiopathic epilepsy in dogs: owners’ perspectives on management with phenobarbitone and/or potassium bromide. Journal of Small Animal Practice 47, 574–581.
Czerwinski, A.L. (1958) Bromide excretion as affected by chloride administration. Journal of American Pharmaceutical Association 47, 467–469.
Dewey, C. (2006) Anticonvulsant therapy in dogs and cats. The Veterinary Clinics of North America - Small Animal Practice 36, 1107–1127.
Dewey, C.W., Ducote, J.M. and Coates, J.R. (1999) Intrarectally administered potassium bromide loading in normal dogs. Journal of Veterinary Internal Medicine 13, 238.
Djuric, M., Kravljanac, R., Kovacevic, G. and Martic, J. (2011) The efficacy of bromides, stiripentol and levetiracetam in two patients with malignant migrating partial seizures in infancy. Epileptic Disorders 13, 22–26.
Dowling, P.M. (1994) Management of canine epilepsy with phenobarbital and potassium bromide. The Canadian Veterinary Journal 35, 724–725.
Ducoté, J.M. (1999) Potassium Bromide. Compendium on Continuing Education for the Practicing Veterinarian 21, 638–639.
Friedlander, W.J. (2000) The rise and fall of bromide therapy in epilepsy. Archives of Neurology 57, 1782–1785.
Gaskill, C.L. and Cribb, A.E. (2000) Pancreatitis associated with potassium bromide/Phenobarbital combination therapy in epileptic dogs. The Canadian Veterinary Journal 41, 555–558.
Gindiciosi, B., Palus, V., Eminaga, S., Villiers, E. and Cherubini, G. (2014) Serum bromide concentrations following loading dose in epileptic dogs. Journal of Small Animal Practice 55, 108–111.
Greenberg, D.M., Aird, R.B. and Boelter, M.D. (1943) A study with radioactive isotopes of the permeability of the blood-cerebrospinal fluid barrier to ions. The American Journal of Physiology 140, 47–64.
Hess, R.S., Kass, P.H., Shofer, F.S., Van Winkle, T.J. and Washabau, R.J. (1999) Evaluation of risk factors for fatal acute pancreatitis in dogs. Journal of the American Veterinary Medical Association 214, 46–51.
Kantrowitz, L.B., Peterson, M.E., Trepanier, L.A., Melián, C. and Nichols, R. (1999) Serum total thyroxine, total triiodothyronine, free thyroxine, and thyrotropin concentrations in epileptic dogs treated with anticonvulsants. Journal of the American Veterinary Medical Association 214, 1804–1808.
Kluger, E.K., Malik, R., Ilkin, W.J., Snow, D., Sullivan, D.R. and Govendir, M. (2008) Serum triglyceride concentration in dogs with epilepsy treated with phenobarbital or with phenobarbital and bromide. Journal of the American Veterinary Medical Association 233, 1270–1277.
Korinthenberg, R., Burkart, P., Woelfle, C., Moenting, J.S. and Ernst, J.P. (2007) Pharmacology, efficacy, and tolerability of potassium bromide in childhood epilepsy. Journal of Child Neurology 22, 414–418.
Locock, C. (1857) Discussion of paper by Sieveking: analysis of 52 cases of epilepsy observed by the author. The Medical Times and Gazette 1, 524–526.
March, P.A., Podell, M. and Sams, R.A. (2002) Pharmacokinetics and toxicity of bromide following high-dose oral potassium bromide administration in healthy Beagles. Journal of Veterinary Pharmacology and Therapeutics 25, 425–432.
Millns, J.L. and Rogers, R.S. 3rd (1978) Furosemide as an adjunct in the therapy of bromism and bromoderma. Dermatologica 156, 111–119.
Nichols, E.S., Trepanier, L.A. and Linn, K. (1996) Bromide toxicosis secondary to renal insufficiency in an epileptic dog. Journal of the American Veterinary Medical Association 208, 231–233.
Palmer, J.W. and Clarke, H.T. (1933) The elimination of bromides from the bloodstream. Journal of Biological Chemistry 99, 435–444.
Paull, L.C., Scott-Moncrieff, J.C., DeNicola, D.B., Glickman, N., Refsal, K.R. and Glickman, L.T. (2003) Effect of anticonvulsant dosages of potassium bromide on thyroid function and morphology in dogs. Journal of The American Animal Hospital Association 39, 193–202.
Pearce, L.K. (1990) Potassium bromide as an adjunct to phenobarbital for the management of uncontrolled seizures in dogs. Progress in Veterinary Neurology 1, 95–101.
Pedersoli, W.M. (1980) Serum bromide concentrations during and after halothane anesthesia in dogs. American Journal of Veterinary Research 41, 77–80.
Podell, M. (1998) Antiepileptic drug therapy. Clinical Techniques in Small Animal Practice 13, 185–192.
Podell, M. (2005) How do I treat status epilepticus. In: Proceedings of the 23rd Annual American College of Veterinary Internal Medicine Forum, Baltimore, Maryland, pp. 366–368.
Podell, M. and Fenner, W.R. (1993) Bromide therapy in refractory canine idiopathic epilepsy. Journal of Veterinary Internal Medicine 7, 318–327.
Podell, M. and Fenner, W.R. (1994) Use of bromide as an antiepileptic drug in dogs. Compendium on Continuing Education for the Practicing Veterinarian 16, 767–774.
Podell, M., March, P.A. and Sams, R. (2000) Successful loading by continuous intravenous administration of sodium bromide in the dog. Proceedings of the 18th Annual American College of Veterinary Internal Medicine Forum, Omnipress, Madison, Wisconsin, p. 718.
Rosenblum, I. (1958) Bromide intoxication. I. Production of experimental intoxication in dogs. The Journal of Pharmacology and Experimental Therapeutics 122, 379–385.
Rossmeisl, J.H. and Inzana, K.D. (2009) Clinical signs, risk factors, and outcomes associated with bromide toxicosis (bromideomism) in dogs with idiopathic epilepsy. Journal of the American Veterinary Medical Association 234, 1425–1431.
Rossmeisl, J.H. Jr, Zimmerman, K., Inzana, K.D. and Higgins, M.A. (2006) Assessment of the use of plasma and serum chloride concentrations as indirect predictors of serum bromide concentrations in dogs with idiopathic epilepsy. Veterinary Clinical Pathology 35, 426–433.
Schwartz-Porsche, D. and Jürgens, U. (1991) Efficacy of potassium bromide against canine epilepsy unresponsive to other treatments. Tierärztl Praxis 19, 395–401.
Schwartz-Porsche, D., Jurgens, U., May, T., Gerhardt, M., Boenigk, H.E. and Krebs, B. (1990) Pharmacokinetics of bromide and bromide therapy in canine epilepsy. Proceedings of the 4th Annual European Society of Veterinary Neurology, Bern, Switzerland, pp. 32–34.
Shaw, N., Trepanier, L.A., Center, S.A. and Garland, S. (1996) High dietary chloride content associated with loss of therapeutic serum bromide concentrations in an epileptic dog. Journal of the American Veterinary Medical Association 208, 234–236.
Shihab, N., Bowen, J. and Volk, H.A. (2011) Behavioral changes in dogs associated with the development of idiopathic epilepsy. Epilepsy and Behaviour 21(2), 160–167.
Steiner, J.M., Xenoulis, P., Anderson, J.A., Barr, A.C. and Williams, D.A. (2008) Serum pancreatic lipase immunoreactivity concentration in dogs treated with potassium bromide and/or phenobarbital. Veterinary Therapeutics 9, 37–44.
Steinmetz, S., Tipold, A., Bilzer, T. and Schenk, H.C. (2012) Transient neuromyopathy after bromide intoxication in a dog with refractory epilepsy. Journal of Veterinary Internal Medicine 26, 839.
Suzuki, S., Kawakami, K., Nakamura, F., Nishimura, S., Yagi, K. and Seino, M. (1994) Bromide, in the therapeutic concentration, enhances GABA-activated currents in cultured neurons of rat cerebral cortex. Epilepsy Research 19, 89–97.
Swell, A.J. (1907) The Dog’s Medical Dictionary. EP Dutton, New York.
Trepanier, L. (1995) Use of bromide as an anticonvulsivant for dogs with epilepsy. Journal of the American Veterinary Medical Association 207, 163–166.
Trepanier, L.A. and Babish, B.J. (1995a) Pharmacokinetics properties of bromide in dogs after the intravenous and oral administration of single doses. Research in Veterinary Science 58, 248–251.
Trepanier, L.A. and Babish, B.J. (1995b) Effect of dietary chloride on the elimination of bromide by dogs. Research in Veterinary Science 58, 252–255.
Trepanier, L.A., Van Schoick, A., Schwark, W.S. and Carrillo, J. (1998) Therapeutic serum drug concentrations in epileptic dogs treated with potassium bromide alone or in combination with other anticonvulsants: 122 cases (1992–1996). Journal of the American Veterinary Medical Association 213, 1449–1453.
Van Dyke, H.B. and Hastings, A.B. (1931) Studies of bromide distribution in the blood II. The distribution of bromides and chlorides in the blood of dogs following oral administration of sodium bromide. Journal of Biological Chemistry 92, 27–32.
Vobecký, M., Pavelka, S. and Babicky, A. (2005) Bromide transfer through mother’s milk and its impact on the suckling rat. Biological Trace Element Research 103, 37–48.
Volk, H.A., Chandler, K.E., Cappello, R. and Cherubini, G.B. (2006) New insights into efficacy and side effects of potassium bromide in epileptic cats. Journal of Veterinary Internal Medicine 20, 780.
Wagner, S.O. (2001) Lower airway disease in cats on bromide therapy for seizures (abstr). In: Proceedings. 19th Annual American College of Veterinary Intern Med Forum 562.
Wallace, G.B. and Brodie, B.B. (1939) The distribution of administered bromide in comparison with chloride and its relation to body fluids. Journal of Pharmacology and Experimental Therapeutics 65, 214–219.
Wolf, R.L. and Eadie, G.S. (1950) Reabsorption of bromide by the kidney. The American Journal of Physiology 163, 436–441.
Woody, R.C., Turley, C.P. and Bromideewster, M.A. (1990) The use of serum electrolyte concentrations determined by automated analyzers to indirectly quantitate serum bromide concentration. Therapeutic Drug Monitoring 12, 490–492.
Yohn, S.E., Morrison, W.B. and Sharp, P.E. (1992) Bromide toxicosis (bromism) in a dog treated with potassium bromide for refractory seizures. Journal of the American Veterinary Medical Association 201, 468–470.
15 Zonisamide
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Zonisamide (ZNS) (1,2-benzisoxazole-3methanesulfonamide) is a synthetic sulfonamidebased anti-epileptic medication (Fig. 15.1), which is structurally unrelated to all other available anti-epileptic medications. It has been available in Japan since 1989 and in South Korea since 1992, but was only licensed in the USA and in Europe in 2000 and 2005, respectively. While the US and European approval is limited to use as an adjunctive treatment of focal seizures in adult humans, the Asian licence includes utilization as mono- and adjunctive therapy for focal and generalized seizures in children and adults. Several studies have indicated that ZNS is well-tolerated and efficacious as first-line or adjunctive anti-epileptic medication in epileptic people with focal seizures and as adjunctive treatment for a wide range of generalized epilepsies (Brodie, 2006; Baulac and Leppik, 2007; Coppola et al., 2009; Shinnar et al., 2009; Lee et al., 2010; Helmstaedter et al., 2011; Holder and Wilfong, 2011; Baulac et al., 2012; Carmichael et al., 2013; Guerrini et al., 2014). ZNS has been shown to decrease appetite and induce weight loss particularly in overweight epileptic people, and therefore may represent a preferable therapeutic option in this patient group (Wellmer et al., 2009; Kim et al., 2012). In addition to its efficacy in treating different seizure types, ZNS may be efficacious in treating various neurological and psychiatric diseases including migraine (Drake et al., 2004), neuropathic pain (Guay, 2003), essential tremor (Bermejo et al., 2008), impulse control disorders (Bermejo and Velasco, 2008) and Parkinson’s disease (Bermejo and Anciones, 2009).
ZNS is generally well tolerated and has a favourable pharmacokinetic profile that permits twice-daily administration and achievement of steady-state plasma concentrations approximately 4 days in dogs and 7 days in cats after treatment initiation or dose change (Dewey et al., 2004; Boothe and Perkins, 2008; Hasegawa et al., 2008; Fukunaga et al., 2010).
ZNS efficacy in reducing seizures has been demonstrated in animal models of both focal and generalized epilepsy (Masuda et al., 1979; Ito et al., 1980, 1986; Wada et al., 1990a, b). Clinical studies on efficacy are limited in dogs and lacking in cats. Reported efficacy (in decreasing seizure frequency by ³50%) in idiopathic epileptic dogs ranges from 58 to 80% when used as adjunctive treatment and is 60% as monotherapy (Dewey et al., 2004; Von Klopman et al., 2007; Chung et al., 2012).
Further studies are needed to evaluate the safety and clinical efficacy of ZNS in cats.
Mechanism of Action
ZNS anticonvulsant activity, which was fortuitously discovered, has been proven in a
number of animal and in vitro experimental seizure models (Masuda et al., 1979; Ito et al., 1980, 1986; Wada et al., 1990a, b; Baulac, 2006; Biton, 2007). Current evidence suggests that ZNS has multiple and complementary mechanisms of action, which likely contribute to its efficacy across a broad range of epilepsy types (Mori et al., 1998; Sobieszek et al., 2003; Leppik, 2004; Baulac, 2006; Biton, 2007). These include:
1. Neuronal membrane stabilization and suppression of neuronal hypersynchronization by inhibition of neuronal voltage-gated sodium channels and inhibition of low-threshold T-type voltage-gated calcium channels (Fig. 15.2).
O N
O S O

NH2
In addition, ZNS has a modest inhibitory effect on carbonic anhydrase, which is thought to be 100 to 200 times less potent than that of acetazolamide. This mechanism does not seem to contribute to ZNS anti-epileptic actions (Masuda and Karasawa, 1993).
Metabolism and Pharmacokinetics
ZNS is rapidly absorbed from the gastrointestinal tract, and peak serum concentrations are achieved 2.5 to 6 h after oral dosing in dogs (Matsumoto et al., 1983; Boothe and Perkins, 2008; Orito et al., 2008). In humans, food can reduce the rate (from 2–5 h to 4–6 h), but not the extent, of absorption. In dogs, ZNS oral bioavailability is 68 ± 12% and approximately 40% of ZNS is protein bound (Boothe and Perkins, 2008). Pharmacokinetic parameters of ZNS in dogs are presented in Table 15.1.

Fig. 15.2. Neuronal receptor targets for zonisamide.
ZNS concentration and disposition differ among blood compartments (i.e. serum, plasma, whole blood, red blood cells) in dogs (Table 15.1), as well as rats and humans (Matsumoto et al., 1983; Boothe and Perkins, 2008). ZNS has a high binding affinity for red blood cells (RBC) and accumulates within these cells. Therefore ZNS RBC and whole-blood concentration and disposition are similar to each other but different from serum and plasma (Matsumoto et al., 1983; Boothe and Perkins, 2008). After a single IV or oral dose, ZNS maximum concentration (Cmax) in serum or plasma is lower than RBC. After a single oral dose the time to reach C (T) is shorter in serum
maxmax
compared to RBC. After a single IV or oral dose, apparent volume of distribution (Vd) is greater in serum and plasma compared to whole blood and RBC, and elimination half-life (T1/2) is more rapid from serum and plasma compared to whole blood and RBC. Clearance of total drug is greater in serum and plasma compared to whole blood and RBC (Boothe and Perkins, 2008). After multiple oral dosing of ZNS at 10.3 mg/kg every 12 h, steady-state concentrations are reached after approximately 4 days (Boothe and Perkins, 2008). At steady-state, the percentage fluctuation between Cmax and Cmin during a 12 h dosing interval is approximately 10%. Elimination T1/2 is more rapid from serum and plasma compared to whole blood and RBC (Boothe and Perkins, 2008).
One study demonstrated that plasma ZNS concentrations after dosing of 10 and 30 mg/ kg/day achieve steady-state concentrations and are proportional to the dose. In contrast, plasma ZNS concentrations are disproportionately high after dosing at 75 mg/kg/day and do not reach steady-state during 52 weeks of administration (Walker et al., 1988).
ZNS readily penetrates across lipid membranes and the blood-brain barrier. ZNS concentration remains higher in the CNS than in serum or plasma. Rat studies indicate that approximately 50% of ZNS reaches the CNS from RBC during a single trans-capillary passage and that there is little efflux of ZNS from the brain (Cornford and Landon, 1985). ZNS concentration in the brain is 25% higher than in plasma within 12 h of oral administration (Matsumoto et al., 1983).
Approximately 10% of ZNS is excreted unchanged in the urine and the remaining 90% is hepatically metabolized before being excreted in the urine (approximately 80%) and in faeces (approximately 10%) (Matsumoto et al., 1983; Boothe and Perkins, 2008). The mechanism of hepatic metabolism of ZNS in dogs and in cats has not been fully elucidated yet.
In humans, ZNS is primarily (50%) metabolized by CYP3A4 (and to a lesser extent by CYP2C19 and CYP3A5) and subsequently conjugated to a glucuronide metabolite (Nakasa et al., 1998). A smaller proportion (20%) of ZNS inactivation occurs by N-acetylation. Approximately 15–30% of ZNS is excreted unchanged in the urine (Shan et al., 2002). In rats, ZNS metabolism seems to occur primarily by N-acetylation and glucuronidation (Stiff and Zemaitis, 1990). N-acetylation and glucuronidation metabolism are deficient in dogs and in cats.
ZNS pharmacokinetics in cats has been reported in one study including five healthy cats administered a single dose of ZNS at 10 mg/kg (Hasegawa et al., 2008). Median maximum plasma concentration (Cmax) was 13.1 µg/ml (range 10.1–14.3 µg/ml), time to maximum plasma concentration (Tmax) was 4 h (range 2–8 h), plasma apparent elimination half-life (T1/2) was 33 h (range 21.3–44.8 h) and areas under the curves (AUCs) were 720.3 µg/ml/h, (range 581.9–753.5 mg/ml/h) (Hasegawa et al., 2008). In healthy cats administered ZNS at 20 mg/kg/ day for 9 weeks there were no significant differences among the plasma ZNS concentrations measured on the second, fourth and eighth weeks. Mean peak (3 h after ZNS administration) and trough concentrations were 59 and 46 µg/ml, respectively (Hasegawa et al., 2008).
Pharmacokinetic Interactions and Adverse Reactions
In humans, ZNS is metabolized predominantly by hepatic CYP3A4, and co-administration
Table 15.1. Pharmacokinetic parameters of zonisamide after intravenous and oral administration in healthy dogs (modified from Matsumoto et al., 1983; Boothe and Perkins, 2008; Orito et al., 2008).
Pharmacokinetic parameters
Dose Blood Reference administered compartment F (%) Vd (l/kg) Cl (ml/h/kg) AUC (h·µg/ml) Cmax (µg/ml) Tmax (h) T½ (h) MRT (h)
| Boothe and | 6.9 mg/kg IV | Serum | NA | 1193 ± 510 | 54 ± 23 | 145 ± 54 | 8.4 ± 2.7 | 0.93 ± 1.0 | 16.4 ± 7.8 | 22.4 ± 7.8 |
| Perkins, | once | Plasma | NA | 1096 ± 252 | 58 ± 11 | 122 ± 33 | 8 ± 1.1 | 0.44 ± 0.54 | 12.9 ± 3.6 | 19.5 ± 4.87 |
| 2008 | Whole blood | NA | 673 ± 62 | 11.4 ± 2.93 | 684 ± 257 | 12.6 ± 1.15 | 0.49 ± 0.76 | 44 ± 13.8 | 63 ± 19.6 | |
| RBC | NA | 379 ± 43 | 5.06 ± 1.8 | 1587 ± 630 | 22.5 ± 4.0 | 0.63 ± 0.81 | 57.4 ± 21.7 83.8 ± 30.5 | |||
| 10.3 mg/kg | Serum | NA | 729 ± 152 | 30.5 ± 8.5 | 355 ± 86 | 13.2 ± 2.0 | 2.5 ± 0.65 | 17.4 ± 4.9 | 26.8 ± 6.6 | |
| PO once | Plasma | 68 ± 12 | 765 ± 150 | 32 ± 7.3 | 347 ± 80 | 14.4 ± 2.3 | 2.75 ± 1.25 | 17.2 ± 3.6 | 26.9 ± 4.6 | |
| Whole blood | NA | 596 ± 180 | 9.6 ± 2.8 | 1107 ± 315 | 19 ± 2.3 | 3.5 ± 1.04 | 46 ± 17.8 | 67 ± 24 | ||
| RBC | NA | 0.40 ± 0.11 | 0.01 ± 0.001 | 3012 ± 1733 | 29 ± 4.4 | 4.3 ± 1.3 | 91 ± 70 | 132 ± 99 | ||
| 10.3 mg/kg | Serum | NA | NA | NA | 2239 ± 851 | 52 ± 8.7 | NA | 21.4 ± 5.4 | NA | |
| PO q12h for | Plasma | NA | NA | NA | 2662 ± 934 | 58 ± 13 | NA | 23.5 ± 5.8 | NA | |
| 8 weeks | Whole blood | NA | NA | NA | 2991 ± 600 | 57 ± 15 | NA | 31 ± 4.3 | NA | |
| RBC | NA | NA | NA | 3683 ± 101 | 55 ± 22 | NA | 37 ± 12.2 | NA | ||
| Matsumoto | 20 mg/kg PO | Plasma | NA | 0.9 | NA | 486 | 20 | 3 | 15 | NA |
| et al., 1983 | once | RBC | NA | NA | 23 | 7.5 | 42 | NA | ||
| Orito et al., | 5 mg/kg PO | Serum | NA | NA | NA | NA | 3.8 | 6 | 13 | NA |
| 2008 | once | |||||||||
PO, per os; IV, intravenously; F, bioavailability; Vd, volume of distribution; Cl, clearance; AUC, area under the curve; C, peak concentration; T, time to peak concentration; T½,
maxmax
elimination half-life; MRT, mean residence time
with other medications that induce or inhibit CYP3A4 may change ZNS’s pharmacokinetics. Co-administration with PB, a CYP3A4 inducer, increases ZNS clearance by about 50% and decreases ZNS elimination half-life (Zaccara and Specchio, 2009). An in vitro study in human hepatic microsomes has shown that CYP inhibitors such as ketoconazole, cyclosporin A and miconazole decrease ZNS clearance by 31%, 23% and 17%, respectively (Nakasa et al., 1998).
In dogs, repeated PB administration enhances CYP3A activity (Hojo et al., 2002) but CYPs involved in ZNS metabolism have not been established yet. However, it has been shown that concurrent administration of PB alters ZNS pharmacokinetics. Repeated oral administration of PB (5 mg/kg every 12 h for 30–35 days) decreased the bioavailability, maximum serum concentration, area under the serum concentration versus time curve and apparent elimination half-life, and increased the total clearance of ZNS (Orito et al., 2008). Time to maximum serum concentration and volume of distribution were not affected by concurrent PB administration. ZNS pharmacokinetic parameters were restored to the same values as before PB administration 12 weeks after the discontinuation of PB (Orito et al., 2008).
ZNS does not appear to affect its own metabolism or disposition of other medications as it has not been shown to induce or inhibit hepatic CYP450 isoenzymes (Masuda et al., 1979; Mimaki, 1998).
ZNS is a weak carbonic anhydrase inhibitor and therefore caution is warranted when it needs to be administered concurrently with other carbonic anhydrase inhibitors.
Dose-related adverse effects of ZNS
The most frequently observed adverse effects of ZNS in humans include somnolence, dizziness, decreased appetite or anorexia and nausea (Leppik, 2006; Zaccara and Specchio, 2009; Zaccara et al., 2011). Other reported adverse effects include fatigue, headache, neuropsychiatric symptoms (e.g. depression, aggressive behaviour, psychosis and irritability), cognitive disturbances, diplopia, weight loss, diarrhoea, ataxia, oligohydrosis, urolithiasis (calcium phosphate or calcium oxalate) and nephrolithiasis (Faught, 2004; Leppik, 2006; Wroe, 2007; Shinnar et al., 2009; Zaccara and Specchio, 2009; Stephen et al., 2010; White et al., 2010). Gradual titration to final maintenance dosage can reduce the incidence of several of the above dose-dependent CNS adverse effects (Zaccara et al., 2011).
In dogs, reported ZNS adverse effects include (Dewey et al., 2004; Von Klopman et al., 2007; Chung et al., 2012):
The prevalence of these adverse effects varies among studies from 10% (1/10 dogs) (Chung, 2012) to 55% (6/11 dogs) (Von Klopman et al., 2007). While in some dogs ataxia and sedation were transient and required no dosage change (Dewey et al., 2004; Von Klopman et al., 2007), in other individuals a dose reduction was necessary (Dewey et al., 2004). Sedation, vomiting and inappetence resolved in one dog following discontinuation of ZNS (Chung et al., 2012). As in humans, gradual titration to final maintenance dosage may help to reduce the incidence and severity of these adverse effects.
Administration of ZNS at 10 mg/kg daily for 9 weeks in two cats did not result in any observable adverse effects. Anorexia, diarrhoea, vomiting, somnolence and ataxia have been observed in three of six healthy adult cats that were administered ZNS at 20 mg/ kg/day for 9 weeks (Hasegawa et al., 2008). The plasma concentrations of ZNS in the three cats with adverse effects (mean trough concentrations 73.8, 49.9 and 41.9 µg/ml) were significantly higher than those in the three cats without adverse effects (mean trough concentrations 41.3, 38.1 and 32.8 µg/ml). In addition, no significant changes were identified in body weight as well as haematology and serum biochemistry evaluated before and after the 9-week administration of ZNS at 20 mg/kg/day (Hasegawa et al., 2008). In a review article it is briefly mentioned that ZNS had to be discontinued due to anorexia in one of two cats in which it was administered (unknown dosage) (Dewey, 2006). The safety of ZNS in cats needs further assessment.
Suspected idiosyncratic adverse effects of ZNS
Idiosyncratic reactions to ZNS in humans are rare and include cutaneous reactions (urticaroid eruptions to epidermal necrolysis), hepatic toxicity, aplastic anaemia and agranulocytosis (Zaccara et al., 2007, 2011). Patients with a previous allergic episode to sulfonamidecontaining medications are at a higher risk for developing ZNS-induced cutaneous rash (Zaccara et al., 2011).
Keratoconjunctivitis sicca (in one dog) and polyarthropathy (in one dog), which are both potential adverse effects of sulfonamidebased medications, have been reported although a clear cause–effect relationship to ZNS could not be proven (Dewey et al., 2004).
Suspected idiosyncratic adverse effects of ZNS in dogs include:
One of the two dogs with acute hepatopathy developed inappetence and vomiting 10 days after initiation of ZNS at 7.7 mg/kg every 12 h. Haematology and serum biochemistry revealed lymphopenia, eosinopenia, mild thrombocytopenia with platelet clumps, marked increase in ALT (16328 U/l) and AST (5908 U/l) activities, moderate increase in ALP (354 U/l) and GGT (61 U/l) activities and hyperbilirubinaemia (2.3 mg/dl). Urinalysis showed bilirubinuria, 2+ proteinuria, pH 7.5, and crystals consistent with sulfonamide drug-related crystalluria. Prothrombin time (PT) and activated partial thromboplastin time (aPTT) were prolonged beyond measurable range. Abdominal ultrasound did not reveal any abnormalities. Despite discontinuation of ZNS and intensive care the dog deteriorated and required humane euthanasia. Post-mortem examination revealed acute toxic hepatic injury (panlobular hepatic necrosis, hepatic lipidosis and lymphoplasmacytic and neutrophilic hepatitis) with secondary coagulopathy and ischaemic injury to the brain, heart, kidney, colon and pancreas; and myeloid hypoplasia (Miller et al., 2011).
Another dog reported with acute hepatopathy presented with vomiting, inappetence and icterus 3 weeks after initiation of ZNS at
8.3 mg/kg, every 12 h (Schwartz et al., 2011). Serum biochemistry was unremarkable prior to initiation of ZNS and showed markedly elevated hepatic enzyme activities (AST 1275 U/l; ALT 3197 U/l; ALKP 5182 U/l; GGT 23 U/l), increased total bilirubin (4.3 mg/dl) and cholesterol (700 mg/dl) 3 weeks after initiation of ZNS. Abdominal ultrasound did not reveal any abnormalities of the hepatic parenchyma. Discontinuation of ZNS and supportive care resulted in complete clinical recovery with normalization of hepatic parameters in 8 weeks (Schwartz et al., 2011).
The dog that developed renal tubular acidosis had been treated with ZNS at a dosage of 7.9–8.4 mg/kg for 18 months. Physical examination was unremarkable, although the dog was panting persistently and appeared agitated. Serum biochemistry abnormalities included hyperchloraemia (124 mmol/l; normal 107–116 mmol/l), hypernatraemia (150 mmol/l; normal 139–147 mmol/l), hypokalaemia (3.1 mmol/l; normal 3.3–4.6 mmol/l), hypophosphataemia (1.6 mg/dl; normal 2.9–6.2 mg/dl) and low total carbon dioxide (TCO2; 11 mmol/l, reference range 21–28 mmol/l). Serum ZNS concentration was 38 µg/ml 6 h after dosing. Urinalysis showed trace proteinuria. Abdominal ultrasound revealed hyperechogenicity of the inner part of each renal cortex. Discontinuation of ZNS resulted in clinical improvement and resolution of the serum biochemistry abnormalities (Cook et al., 2011).
On the basis of these reports, serum hepatic enzymes, bilirubin, electrolytes, acid-base status and haematology should be assessed before initiation of ZNS and monitored periodically during ZNS treatment. An increase in serum chloride concentration and a decrease in TCO2 should prompt further investigation of renal tubular acidosis (Cook et al., 2011).
ZNS-related laboratory changes
ZNS treatment may affect thyroid function and some clinical laboratory test results. In a pharmacokinetic study of healthy dogs administered ZNS at 10.3 mg/kg PO every 12 h for 8 weeks, mean total T4 decreased below the normal reference range at study end (6 months) compared to baseline. Mean free T4 and TSH decreased and increased at study end compared to baseline, respectively, but remained within reference range. Serum alkaline phosphatase and calcium increased, and serum total protein and albumin decreased, although remained within reference range at study end compared to baseline (Boothe and Perkins, 2008). A small but statistically significant decrease in plasma albumin concentration and an increase in alkaline phosphatase activity have been reported in research dogs administered 75 mg/ kg/day for 52 weeks (Walker et al., 1988).
Dosing and Monitoring Recommendations
The recommended initial dosage of ZNS is 3–7 mg/kg of body weight orally every 12 h in dogs and 7–10 mg/kg of body weight orally every 12 h in dogs co-administered hepatic microsomal enzymes inducers such as PB (Dewey et al., 2004; Boothe and Perkins, 2008; Orito et al., 2008).
The human target range of 10–40 µg/ml can be used as guidance regarding effective concentrations that can be targeted in dogs. Clinical and serum concentration monitoring can be used to establish a therapeutic range for individual patients and adjust oral dosage accordingly.
Serum or plasma ZNS concentration should be monitored 1 week after treatment initiation or dosage adjustment and any time seizure frequency increases. Currently there are no recommendations on optimal timing of blood sampling for ZNS concentration monitoring. The effect of ZNS dosage on timing of blood collection for serum ZNS concentration monitoring is unknown. In a study assessing both trough and peak serum ZNS concentration in 12 epileptic dogs, all but one trough and all peak serum ZNS concentrations were within the target range of 10–40 µg/ml and estimated peak ZNS concentrations were significantly higher than trough concentrations (Dewey et al., 2004). In a recent pharmacokinetic study, fluctuation between peak and trough concentrations (Cmax and Cmin) was 10% at steady-state (Boothe and Perkins, 2008). The authors recommend collection of a trough sample (within 1 h before the next scheduled administration) as this will enable assessment of the lowest concentration that occurs during a dosing interval and facilitate comparison of results of serial samples by maintaining consistency in the time of blood sampling in relation to the time of ZNS administration. In dogs with seizures that are difficult to control (especially dogs administered ZNS concurrently with PB), both a trough and peak (3 h after ZNS administration) sample should be collected in order to investigate the potential role of short disappearance half-life in causing therapeutic failure.
Based on the limited data available (Hasegawa et al., 2008), the proposed ZNS dosage in cats is 5–10 mg/kg every 24 h, although this has not been evaluated in the clinical setting. Serum ZNS target range also has not been established yet in cats.
Efficacy
The efficacy of ZNS as mono- or adjunctive therapy has been demonstrated in several randomized, double-blind, placebo-controlled as well as long-term, open-label studies in humans with focal or generalized epilepsies (Faught et al., 2001; Brodie et al., 2005; Baulac, 2007; Coppola et al., 2009; Shinnar et al., 2009; Zaccara and Specchio, 2009; Lee et al., 2010; Helmstaedter et al., 2011; Holder and Wilfong, 2011; Baulac et al., 2012).
Information on the clinical efficacy of ZNS in epileptic dogs is limited to three small open-label, uncontrolled studies (Dewey et al., 2004; Von Klopman et al., 2007; Chung et al., 2012).
Overall ZNS efficacy (in decreasing seizure frequency by ³50%) in epileptic dogs with generalized seizures has been reported as 58–80% when used as adjunctive anti-epileptic medication (Dewey et al., 2004; Von Klopman et al., 2007) and 60% as monotherapy (Chung et al., 2012). The 80% efficacy refers to the first 4 months of ZNS treatment (Von Klopman et al., 2007).
An open label, non-comparative study including 12 idiopathic epileptic dogs poorly controlled with PB alone or in combination with KBr and/or other anti-epileptic medications and administered ZNS at a mean dosage of 8.9 mg/kg every 12 h, reported a median reduction in seizure frequency (when comparing ³8 weeks before and after treatment initiation) of 84.5% (range 54.8–100%, mean 81.3%) in seven (58%) dogs (Dewey et al., 2004). Two of these seven dogs became seizure free. Concurrently administered anti-epileptic medications including PB, KBr, felbamate or clorazepate could be reduced in dosage or discontinued in all seven responders to ZNS. The mean reduction of PB dosage was 92.2%.The remaining five dogs (42%) experienced a median increase in seizure frequency of 52.6% (range 7.4–2500%). All 12 dogs in this study experienced generalized seizures, two of which also displayed focal seizures (Dewey et al., 2004). The mean and median follow-up times after ZNS administration were 33.5 weeks and 37 weeks, respectively (range 8–71 weeks). In all dogs the oral dosage of ZNS was adjusted in order to achieve serum concentration between 10 and 40 µg/ml. There were no significant differences between the serum ZNS concentrations of responders versus non-responders for either trough or peak values (Dewey et al., 2004).
Another open label, non-comparative study included 11 idiopathic epileptic dogs with generalized seizures poorly responsive to PB and/or KBr and administered ZNS at 10 mg/kg (Von Klopman et al., 2007). ZNS was administered as adjunctive treatment in ten dogs and as monotherapy in one dog that developed PB-induced haematological abnormalities. Seizure frequency during the 4 months before and after ZNS treatment was compared. Eight of the ten dogs (80%) administered ZNS as adjunctive treatment had a median reduction in seizure frequency of 82.7% (range 58–100%) during the 4 months following ZNS treatment initiation. However, seizure frequency increased in three of these eight dogs in the longer term follow-up (7–17 months). The dog on ZNS monotherapy experienced a seizure reduction of 100% with a 17-month follow up. The remaining two dogs included in the study had a seizure reduction of 14% and 25%, respectively. Seizure duration and severity (e.g. single seizures instead of cluster seizures or status epilepticus) decreased in two dogs. The PB and/or KBr dose could be reduced in seven dogs without subsequent impairment of seizure control (Von Klopman et al., 2007).
The only study on the use of ZNS as monotherapy for canine idiopathic epilepsy included ten dogs with generalized-onset seizures receiving ZNS at 5–15 mg/kg orally every 12 h to achieve serum ZNS concentration of 10–40 µg/ml (Chung et al., 2012). Of these dogs, 60% (6/10) had a ³50% reduction in monthly frequency of seizures with a follow-up of 12–36 months. The mean ZNS dosage in these six dogs was 7.92 ± 3.79 mg/kg twice a day. The remaining four dogs had unsatisfactory response (e.g. seizure frequency was unchanged in two dogs and increased in two other dogs). Mean peak (3 h after oral administration) ZNS serum concentrations were 15.24 ± 5.95 µg/ml (range 7.7–24 µg/ml) in the six dogs with favourable response and
22.41 ± 19.69 µg/ml (range 9.3–51.6 µg/ml) in the four dogs with an unsatisfactory response (Chung et al., 2012).
ZNS has been effective in controlling seizure activity in experimental cats (Ito et al., 1980, 1986; Wada et al., 1990a, b), however information on clinical efficacy is very limited and further data are needed before ZNS use can be recommended in cats. In a review article, one author has reported information on the use of ZNS as adjunctive treatment to PB in two epileptic cats (Dewey, 2006). One cat became anorexic, necessitating discontinuation of ZNS. The other cat experienced a substantial reduction in seizure frequency and showed no clinical or laboratory abnormalities attributable to ZNS administration for about 1 year (Dewey, 2006).
Summary Recommendations
References
Baulac, M. (2006) Introduction to zonisamide. Epilepsy Research 68, 2.
Baulac, M. and Leppik, I.E. (2007) Efficacy and safety of adjunctive zonisamide therapy for refractory partial seizures. Epilepsy Research 75, 75–83.
Baulac, M., Brodie, M.J., Patten, A., Segieth, J. and Giorgi, L. (2012) Efficacy and tolerability of zonisamide versus controlled-release carbamazepine for newly diagnosed partial epilepsy: a phase 3, randomised, double-blind, non-inferiority trial. Lancet Neurology 11, 579–588.
Bermejo, P.E. and Anciones, B. (2009) A review of the use of zonisamide in Parkinson’s disease. Therapeutic Advances in Neurological Disorders 2, 313–317.
Bermejo, P.E. and Velasco, R. (2008) Zonisamide in managing impulse control disorders in Parkinson’s disease. Journal of Neurology 255, 167.
Bermejo, P.E., Ruiz-Huete, C., Dorado, R. and Anciones, B. (2008) Zonisamide in refractoryessential tremor. Revista de Neurologia 46, 139–142.
Biton, V. (2007) Clinical pharmacology and mechanism of action of zonisamide. Clinical Neuropharmacology 30, 230–240.
Boothe, D.M. and Perkins, J. (2008) Disposition and safety of zonisamide after intravenous and oral single dose and oral multiple dosing in normal hound dogs. Journal of Veterinary Pharmacology and Therapeutics 31, 544–553.
Brodie, M.J. (2006) Zonisamide as adjunctive therapy for refractory partial seizures. Epilepsy Research 68, 11–16.
Brodie, M.J., Duncan, R., Vespignani, H., Solyom, A., Bitenskkyy, V. and Lucas, C. (2005) Dose-dependent safety and efficacy of zonisamide: a randomized, double-blind, placebo-controlled study in patients with refractory partial seizures. Epilepsia 46, 31–41.
Carmichael, K., Pulman, J., Lakhan, S.E., Parikh, P., Marson, A.G. (2013) Zonisamide addon for drug-resistant partial epilepsy. The Cochrane Database of Systematic Reviews: CD001416.
Chung, J., Hwang, C., Chae, J., Ahn, J., Kim, T., Seo, K., Lee, S. and Youn, H. (2012) Zonisamidemonotherapy for idiopathic epilepsy in dogs. New Zealand Veterinary Journal, 4.
Cook, A.K., Allen, A., Espinosa, D. and Barr, J. (2011) Renal tubular acidosis associated with zonisamide therapy in a dog. Journal of Veterinary Internal Medicine 25, 1454–1457.
Coppola, G., Grosso, S., Verrotti, A., Parisi, P., Luchetti, A., Franzoni, E., Mangano, S., Pelliccia, A., Operto, F.F., Iannetti, P., Curatolo, P., Balestri, P. and Pascotto, A. (2009) Zonisamide in children and young adults with refractory epilepsy: an open label, multicenter Italian study. Epilepsy Research 83, 112–116.
Cornford, E.M. and Landon, K.P. (1985) Blood-brain barrier transport of CI-912: single-passage equilibration of erythrocyte-borne drug. Therapeutic Drug Monitoring 7, 247–254.
Dewey, C.W. (2006) Anticonvulsant therapy in dogs and cats. Veterinary Clinics of North America – Small Animal Practice 36(5), 1107–1127.
Dewey, C.W., Guiliano, R., Boothe, D.M., Berg, J.M., Kortz, J.D., Joseph, R.J. and Budsberg, S.C. (2004) Zonisamide therapy for refractory idiopathic epilepsy in dogs. Journal of the American Animal Hospital Association 40, 285–291.
Drake, M.E. Jr, Greathouse, N.I., Renner, J.B. and Armentbright, A.D. (2004) Open label zonisamide for refractory migraine. Clinical Neuropharmacology 27, 278–280.
Faught, E. (2004) Review of United States and European clinical trials of zonisamide in the treatment of refractory partial-onset seizures. Seizure 13, 59–65.
Faught, E., Ayala, R., Montouris, G.G. and Leppick, I.E. (2001) Randomized controlled trial of zonisamide for the treatment of refractory partial-onset seizures. Neurology 57, 1774–1779.
Fukunaga, K., Saito, M., Muto, M., Yoshioka, R., Mishima, K., Fujiwara, M. and Orito, K. (2010) Steady-state pharmacokinetics of zonisamide in plasma, whole blood, and erythrocytes in dogs. Journal of Veterinary Pharmacology and Therapeutics 33, 103–106.
Guay, D.R. (2003) Oxcarbazepine, topiramate, zonisamide, and levetiracetam: potential use in neuropathic pain. The American Journal of Geriatric Pharmacotherapy 1, 18–37.
Guerrini, R., Rosati, A., Bradshaw, K., Giorgi, L. (2014) Adjunctive zonisamide therapy in the long-term treatment of children with partial epilepsy: Results of an open-label extension study of a phase III, randomized, double-blind, placebo-controlled trial. Epilepsia, 55(4), 568–578.
Hasegawa, D., Kobayashi, M., Kuwabara, T., Ohmura, T., Fujita, M. and Orima, H. (2008) Pharmacokinetics and toxicity of zonisamide in cats. Journal of Feline Medicine and Surgery 10, 418–421.
Helmstaedter, C., Stefan, H. and Witt, J.A. (2011) Quality of life in patients with partial-onset seizures under adjunctive therapy with zonisamide: results from a prospective non-interventional surveillance study. Epileptic Disorders 13, 263–276.
Hojo, T., Ohno, R., Shimodo, M. and Kokue, E. (2002) Enzyme and plasma protein induction by multiple oral administrations of phenobarbital at a therapeutic dosage regimen in dogs. Journal of Veterinary Pharmacology and Therapeutics 25, 121–127.
Holder, J.L. Jr and Wilfong, A.A. (2011) Zonisamide in the treatment of epilepsy. Expert Opinion on Pharmacotherapy 12, 2573–2581.
Ito, T., Hori, M., Masuda, Y., Yoshida, K. and Shimizu, M. (1980) 3-Sulfamoylmethyl-1,2-benzisoxazole, a new type of anticonvulsant drug. Electroencephalographic profile. Arzneimittel-Forschung 30, 603–609.
Ito, T., Hori, M. and Kadokawa, T. (1986) Effects of zonisamide (AD-810) on tungstic acid gel-induced thalamic generalized seizures and conjugated estrogen-induced cortical spike-wave discharges in cats. Epilepsia 27, 367–374.
Kim, D.W., Yoo, M.W. and Park, K.S. (2012) Low serum leptin concentration is associated with zonisamideinduced weight loss in overweight female epilepsy patients. Epilepsy and Behaviour 23, 497–499.
Lee, Y.J., Kang, H.C., Seo, J.H., Lee, J.S. and Kim, H.D. (2010) Efficacy and tolerability of adjunctive therapy with zonisamide in childhood intractable epilepsy. Brain and Development 32, 208–212.
Leppik, I.E. (2004) Zonisamide: chemistry, mechanism of action, and pharmacokinetics. Seizure 13, 5–9.
Leppik, I.E. (2006) Practical prescribing and long-term efficacy and safety of zonisamide. Epilepsy Research Suppl. 2, S17–24.
Leppik, I.E., Willmore, L.J., Homan, R.W., Fromm, G., Oommen, K.J., Penry, J.K., Sackellares, J.C., Smith, D.B., Lesser, R.P. and Wallace, J.D. (1993) Efficacy and safety of zonisamide: results of a multicentre study. Epilepsy Research 14, 165–173.
Masuda, Y. and Karasawa, T. (1993) Inhibitory effect of zonisamide on human carbonic anhydrase in vitro. Arzneimittel-Forschung 43, 416–417.
Masuda, Y., Utsui, Y., Shiraishi, Y., Karasawa, T., Yoshida, K. and Shimizu, M. (1979) Relationships between plasma concentrations of diphenylhydantoin, phenobarbital, carbamazepine, and 3-sulfamoylmethyl1,2-benzisoxazole (AD-810), a new anticonvulsant agent, and their anticonvulsant or neurotoxic effects in experimental animals. Epilepsia 20, 623–633.
Matsumoto, K., Miyazaki, H., Fujii, T., Kagemoto, A., Maeda, T. and Hashimoto, M.H. (1983) Absorption, distribution and excretion of 3-(sulfamoyl[14C]-methyl)1,2-benziosoxazole (AD-810) in rats, dogs and monkeys and of AD-810 in men. Drug Research 33, 961–968.
Miller, M.L., Center, S.A., Randolph, J.F., Lepherd, M.L., Cautela, M.A. and Dewey, C.W. (2011) Apparent acute idiosyncratic hepatic necrosis associated with zonisamide administration in a dog. Journal of Veterinary Internal Medicine 25, 1156–1160.
Mimaki, T. (1998) Clinical pharmacology and therapeutic drug monitoring of zonisamide. Therapeutic Drug Monitoring 20, 593–597.
Mori, A., Noda, Y. and Packer, L. (1998) The anticonvulsant zonisamide scavenges free radicals. Epilepsy Research 30, 153–158.
Nakasa, H., Nakamura, H., Ono, S., Tsutsui, M., Kiuchi, M., Ohmori, S. and Kitada, M. (1998) Prediction of drug-drug interactions of zonisamide metabolism in humans from in vitro data. European Journal of Clinical Pharmacology 54, 177–183.
Orito, K., Saito, M., Fukunaga, K., Matsuo, E., Takikawa, S., Muto, M., Mishima, K., Egashira, N. and Fujiwara, M. (2008) Pharmacokinetics of zonisamide and drug interaction with phenobarbital in dogs. Journal of Veterinary Pharmacology and Therapeutics 31, 259–264.
Schwartz, M., Muñana, K. and Olby, N.J. (2011) Possible drug-induced hepatopathy in a dog receiving zonisamide monotherapy for treatment of cryptogenic epilepsy. The Journal of Veterinary Medical Science 73, 1505–1508.
Shan, J., Shllenberger, K. and Canafox, D.M. (2002) Zonisamide: chemistry, biotrasformation, and pharmacokinetics. In: Levy, R.H., Mattson, R.H., Meldrum B.S. and Perucca, E. (eds) Anitepileptic Drugs, 5th edition. Lippincot, Williams & Wilkins, Philadelphia, Pennsylvania, pp. 873–879.
Shinnar, S., Pellock, J.M. and Conry, J.A. (2009) Open-label, long-term safety study of zonisamide administered to children and adolescents with epilepsy. European Journal of Paediatric Neurology 13, 3–9.
Sobieszek, G., Borowicz, K.K., Kimber-Trojnar, Z., Małtek, R., Piskorska, B. and Czuczwar, S.J. (2003) Zonisamide: a new antiepileptic drug. Polish Journal of Pharmacology 55, 683–689.
Stephen, L.J., Kelly, K., Wilson, E.A., Parker, P. and Brodie, M.J. (2010) A prospective audit of adjunctive zonisamide in an everyday clinical setting. Epilepsy and Behaviour 17, 455–460.
Stiff, D.D. and Zemaitis, M.A. (1990) Metabolism of the anticonvulsant agent zonisamide in the rat. Drug Metabolism and Disposition 18, 888–894.
Von Klopmann, T., Rambeck, B. and Tipold, A. (2007) Prospective study of Zonisamide therapy for refractory idiopathic epilepsy in dogs. Journal of Small Animal Practice 48, 134–138.
Wada, Y., Hasegawa, H., Okuda, H. and Yamaguchi, N. (1990a) Anticonvulsant effects of zonisamide and phenytoin on seizure activity of the feline visual cortex. Brain and Development 12, 206–210.
Wada, Y., Hasegawa, H. and Yamaguchi, N. (1990b) Effect of a novel anticonvulsant, zonisamide (AD-810, CI-912), in an experimental model of photosensitive epilepsy. Epilepsy Research 7, 117–120.
Walker, R.M., DiFonzo, C.J., Barsoum, N.J., Smith, G.S. and Macallum, G.E. (1988) Chronic toxicity of the anticonvulsant zonisamide in beagle dogs. Fundamental and Applied Toxicology 11, 333–342.
Wellmer, J., Wellmer, S. and Bauer, J. (2009) The impact of zonisamide on weight. A clinical study in 103 patients with epilepsy. Acta Neurologica Scandinavica 119, 233–238.
White, J.R., Walczak, T.S., Marino, S.E., Beniak, T.E., Leppik, I.E. and Birnbaum, A.K. (2010) Zonisamide discontinuation due to psychiatric and cognitive adverse events: a case-control study. Neurology 75, 513–518.
Wroe, S. (2007) Zonisamide and renal calculi in patients with epilepsy: how big an issue? Current Medical Research and Opinion 23, 1765–1773.
Yamauchi, T. and Aikawa, H. (2004) Efficacy of zonisamide: our experience. Seizure 13, 41–48.
Zaccara, G. and Specchio, L.M. (2009) Long-term safety and effectiveness of zonisamide in the treatment of epilepsy: a review of the literature. Neuropsychiatric Disease and Treatment 5, 249–259.
Zaccara, G., Franciotta, D. and Perucca, E. (2007) Idiosyncratic adverse reactions to antiepileptic drugs. Epilepsia 48, 1223–1244.
Zaccara, G., Tramacere, L. and Cincotta, M. (2011) Drug safety evaluation of zonisamide for the treatment of epilepsy. Expert Opinion and Drug Safety 10, 623–631.
16 Levetiracetam
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Levetiracetam (LEV) is the S-enantiomer of alpha-ethyl-2-oxo-1-pyrollidine acetamide (C8H14N2O2) and it is structurally related to the nootropic drug piracetam (Fig. 16.1). It is chemically unrelated to and appears to have a different mechanism of action than other currently available anti-epileptic medications (AEMs). Oral LEV was approved by the United States Food and Drug Administration (FDA) in 1999 for adjunctive treatment of refractory focal onset seizures in adults. It was licensed in Europe in September 2000 for the adjunctive treatment of focal onset seizures with or without secondary generalization, and subsequently as monotherapy in 2006. Other approved indications include adjunctive treatment of myoclonic seizures and generalized tonic-clonic seizures associated with idiopathic generalized epilepsy (Berkovic et al., 2007; Noachtar et al., 2008). In humans LEV is generally well-tolerated, safe and efficacious in the treatment of both focal and generalized epilepsies with a positive impact on quality of life (Cereghino et al., 2000; Krauss et al., 2003; Otoul et al., 2005; Abou-Khalil, 2008; Verrotti et al., 2010; Lo et al., 2011; Lyseng-Williamson, 2011; Stephen et al., 2011; Mbizvo et al., 2012; Fang et al., 2014).
LEV has been shown to be a valuable antiepileptic monotherapy option in patients with primary brain tumours due to its efficacy and tolerability (Rossetti 2014). LEV efficacy as AEM has been shown in animal models of chronic epilepsy (De Deyn et al., 1992; Löscher and Hönack, 1993; Gower et al., 1995; Klitgaard et al., 1998; Löscher et al., 1998; Margineanu and Klitgaard, 2003; Niespodziany et al., 2003; Stratton et al., 2003; De Smedt et al., 2005). LEV is well tolerated and generally safe for clinical use in dogs and cats, however data on its clinical efficacy are limited in these species (Steinberg and Faissler, 2004; Dewey et al, 2005; Bailey et al., 2008; Volk et al., 2008; Hardy et al., 2012; Muñana et al., 2012).
Similar to some of the other AEMs such as gabapentin and pregabalin, LEV has also been shown to have antinociceptive effects in various animal models of pain (Ardid et al., 2003; Archer et al., 2007; Ozcan et al., 2008; Sliva et al., 2008; Micov et al., 2010).
LEV has also neuroprotective properties and may minimize seizure and acute ischaemiainduced brain damage (Hanon and Klitgaard, 2001; Rekling, 2003; Christensen et al., 2010; Belcastro et al., 2011; Shetty 2013; Komur et al., 2014). LEV is available as immediate and extended release tablets, oral solution and an intravenous formulation.
Mechanism of Action
The mechanism of action of LEV is not completely understood, but it is thought to act by
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
binding to the synaptic vesicle 2A (SV2A) protein on the presynaptic terminal, and modulating synaptic vesicle fusion and neurotransmitter release (Fig. 16.2) (Lynch et al., 2004; Yang et al., 2007). Other mechanisms that may contribute to LEV anti-epileptic activity include inhibition of the Na+-dependent Cl−/HCO3− exchange (Leniger et al., 2004), modulation of K+ and N-type high-voltageactivated Ca2+ channels (Niespodziany et al, 2001; Lukyanetz et al., 2002; Madeja et al., 2003; Pisani et al., 2004), reduction of glutamate release by modulation of the presynaptic P/Q-type voltage-dependent Ca2+ channels (Lee et al., 2009), opposition of allosteric inhibition of GABA and glycine-gated currents (Rigo et al., 2002) and antagonism of
CH2CH3
H
CONH2 N
 O
O
neuronal hypersynchronization (Margineanu and Klitgaard, 2000). In addition, a recent study suggests that LEV may reduce the spread of excitation elicited by seizures within the astroglial functional syncytium, with stabilizing consequences for neuronal–glial interactions (Stienen et al., 2011). Unlike other AEMs, LEV does not seem to inhibit voltage-gated Na+ channels, T-type calcium channels (Zona et al., 2001), nor modulate GABA receptors (Margineanu and Klitgaard, 2003).
Metabolism and Pharmacokinetics
LEV has a favourable pharmacokinetic profile. It is relatively rapidly and extensively absorbed both parenterally and orally with co-ingestion of food slowing the rate but not the extent of absorption (Beasley, 2012). LEV is <10% protein bound, readily crosses the blood-brain barrier, and follows linear and time-independent kinetics (Benedetti et al., 2004; Patsalos, 2004). In rats, the half-life of LEV in cerebrospinal fluid was significantly longer and approximately twice that of the plasma half-life (Doheny et al., 1999). LEV is
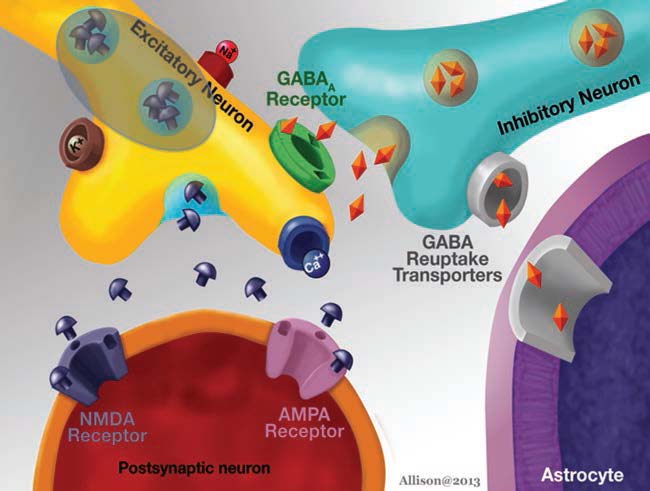
Fig. 16.2. Neuronal receptor targets for levetiracetam.
Levetiracetam 427
primarily (89% in dogs) excreted in the urine (Benedetti et al., 2004) with approximately 50–62% excreted unchanged and the remaining fraction being metabolized primarily through enzymatic hydrolysis by hydrolases, amidases and b-esterases in the bloodstream, liver and other tissues, although a portion of LEV also appears to undergo oxidation (Isoherranen et al., 2001; Benedetti et al., 2004; Patsalos, 2004). LEV is not metabolized by the cytochrome P450 (CYP450) pathway in the liver and autoinduction does not occur (Benedetti et al., 2004). Renal elimination occurs primarily by glomerular filtration. Renal clearance of LEV is similar in dogs and humans, being 0.69 ± 0.16 ml/min/kg (Isoherranen et al., 2001), correlates with creatinine clearance and is progressively reduced in patients with increasing severity of renal dysfunction (Patsalos, 2004).
Pharmacokinetic parameters of LEV in healthy dogs and in cats are presented in Tables 16.1 and 16.2, respectively. Some of the differences among studies may be related to methodology variations. For example, LEV infusion rates and initial blood sampling times were 2 and 2 min, respectively, in one study (Dewey et al., 2008) and 5 and 15 min, respectively, in another study on pharmacokinetics of IV LEV in healthy dogs (Patterson et al., 2008). LEV concentrations were assessed by high-performance liquid chromatography (HPLC) analysis in most studies (Bailey et al., 2008; Dewey et al., 2008; Patterson et al., 2008; Moore et al., 2010; Carnes et al., 2011; Platt et al, 2011), although enantioselective gas-chromatography was used by others (Isoherranen et al., 2001).
Administration of multiple LEV oral doses over 7 days did not alter LEV pharmacokinetics significantly when compared to single dosing (Moore et al., 2010). A diurnal variation in LEV plasma concentrations has been reported with mean morning trough LEV concentration (18.42 ± 5.16 mg/ml) being significantly higher than mean afternoon trough concentration (12.57 ± 4.34 mg/ml) (Moore et al., 2010). It has been suggested that this difference may be due to diurnal variations in glomerular filtration rate (Moore et al., 2010).
Subcutaneous (SC) administration of LEV at 60 mg/kg resulted in plasma LEV concentration above the proposed human therapeutic range of 5–46 mg/ml (Folland and Moriarty, 2002; Juenke et al., 2002; Leppik et al., 2002; Johannessen et al., 2003) within 15 minutes of administration, and remained above the range for at least 7 hours.. Mean plasma LEV concentration was 65.2 ± 29.5, 114.5 ± 10.5 and 84.9 ± 20.6 mg/ml at 15, 120 and 420 min, respectively. Administration of SC LEV was well tolerated (Hardy et al., 2011).
Rectal administration of 40 mg/kg of parenteral levetiracetam resulted in a serum concentration of 15.3 ± 5.5 mg/ml (range 8.5–
22.5 mg/ml) at 10 minutes. The serum LEV concentration remained elevated and exceeded 5 mg/ml in all dogs during the 9 hour sampling period after LEV administration (Peters 2014). Mean C, T and AUC for all dogs
maxmax
are indicated in table 16.1. Dogs with faeces in the rectum at the time of LEV administration had significantly lower mean Cmax values
(26.7 ± 3.4 mg/ml) compared with those without (45.2 ± 4.4 mg/ml). Mild sedation was observed between 60 and 90 minutes after LEV administration without other adverse effects noted.
The pharmacokinetics of extended release (XR) oral LEV has recently been evaluated (Beasley and Boothe, 2012) and compared to immediate release (IRe) oral formulation in healthy dogs (Platt, 2011). The AUC associated with XR was 230 mg/ml/h, 5.14 times higher than that with IRe (44.8 mg/ml/h). The absorption half-life of XR was 3.2 h, 7.75 times higher than with IRe (0.41 h). The elimination half-life was 3.13 h with XR and 2.19 h with IRe, a 1.43fold difference. The Tmax associated with XR was 5.01 h, 4.21 times higher than that associated with IRe (1.22 h). The Cmax associated with XR was 18.5 mg/ml, 1.92 times higher than that associated with IRe (9.62 mg/ml). The plasma concentration of LEV was not detectable at 8 h after administration of IRe LEV, whereas it was greater than 10 mg/ml at 8 h after XR LEV administration (Platt, 2011). Administration of XR LEV with food significantly increased bioavailability and time to maximum concentration compared to those in fasted dogs, although half-life was not significantly affected (Beasley and Boothe, 2012).
One study on pharmacokinetics of IV LEV administered at 30 or 60 mg/kg in dogs
Table 16.1. Pharmacokinetic parameters of LEV after intravenous, intramuscular, oral and rectal administration in healthy dogs (Isoherranen et al., 2001; Dewey, 2008; Patterson, 2008; Moore, 2010; Peters, 2014) (data is presented as mean ± standard deviation when available).
Pharmacokinetic parameters
Reference Dose administered F (%) Vd (l/kg) Cl (ml/kg/min) AUC (h•mg/ml) C (mg/ml) T (h) T½ (h) MRT (h)
maxmax
| Isoherranen et al., | 20 mg/kg IV once | NA | 0.5 ± 0.1 | 1.5 ± 0.3 | NA | NA | NA | 3.6 ± 0.8 | 5.0 ± 1.2 |
| 2001 | |||||||||
| Dewey, 2008 | 60 mg/kg IV once | NA | 0.48 ± 0.08 | 1.4 ± 0.28 | 768 ± 179 | 254 ± 81 | NA | 4.0 ± 0.82 | 6.0 ± 0.9 |
| Patterson, 2008 | 20 mg/kg IV once | NA | 0.55 | 2.1 ± 0.3 | 166 ± 27 | 37 ± 5 | NA | 3 ± 0.3 | NA |
| 20 mg/kg IM once | 113 | 0.6 | 2.3 ± 0.3 | 176 ± 20 | 30 ± 3 | 0.7 ± 0.3 | 3 ± 0.4 | NA | |
| 20 mg/kg PO once | 100 | 0.59 | 2.1 ± 0.3 | 167 ± 23 | 30 ± 4 | 1.4 ± 0.5 | 3 ± 0.3 | NA | |
| Moore, 2010 | 20 mg/kg PO once | NA | NA | NA | 268.52 ± 56.33 | 59.91 ± 11.54 | 0.62 | 2.87 ± 0.21 | NA |
| 20 mg/kg PO q8h for 7 d | NA | NA | NA | 289.31 ± 51.68 | 52.41 ± 10.08 | 1 | 3.59 ± 0.82 | NA | |
| Peters, 2014 | 40 mg/kg rectally | NA | NA | NA | 222 ± 72 | 36.0 ± 10.7 | 1.7 ± 0.5 | NA | NA |
PO= per os; IV= intravenously; d=days; F=bioavailability, Vd=volume of distribution; Cl= clearance; AUC= area under the curve; C = maximum concentration; T = time to maximum
maxmax
concentration; T½ = elimination half-life; MRT= mean residence time
Table 16.2. Pharmacokinetic parameters of LEV in cats with suspected idiopathic epilepsy (Bailey et al., 2008) and in healthy cats (Carnes et al., 2011).
Pharmacokinetic parameters
Dose Cl (ml/kg/ Cl/F AUC Cmin Reference administered F (%) Vd (l/kg) Vd/F (l/kg) min) (ml/kg/h) (h•mg/ml) Cmax (mg/ml) (mg/ml) Tmax (h) T½ (h) MRT (h)
| Bailey | 20 mg/kg | Median | NA | NA | 0.5 | NA | 1.72 | 122.7 | 25.5 | 8.3 | 2 | 2.9 | 3.9 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| et al., | PO q8h | Range | NA | NA | 0.38–1.0 | NA | 0.63–3.68 | 85.0–186.2 | 16.4–39.5 | 2–15 | 2.0–6.0 | 1.9–9.6 | 3.6–4.9 | |||||||||||||
| 2008 | for 7 days | |||||||||||||||||||||||||
| Carnes | 20 mg/kg IV | Mean | NA | 0.52 ± 0.09 | NA | 2.0 ± 0.60 | NA | 179.9 ± 38.6 37.52 ± 6.79 | NA | NA | 2.86 ± 0.65 4.57 ± 0.94 | |||||||||||||||
| et al., | once | ± pseudo | ||||||||||||||||||||||||
| 2011 | SD | |||||||||||||||||||||||||
| Median | NA | 0.51 | NA | 1.7 | NA | 191.3 | 37.64 | NA | NA | 3.1 | 4.77 | |||||||||||||||
| Range | NA | 0.33–0.64 | NA | 1.5–3.4 | NA | 96.7–217.1 28.05–51.86 | NA | NA | 2.07–4.08 3.09–6.09 | |||||||||||||||||
| 20 mg/kg | Mean | 102 ± 39 | NA | NA | NA | NA | 172.9 ± 38.6 25.54 ± 7.97 | NA | 1.67 ± 1.73 2.95 ± 0.95 5.65 ± 1.25 | |||||||||||||||||
| PO once | ± pseudo | |||||||||||||||||||||||||
| SD | ||||||||||||||||||||||||||
| Median | 0.94 | NA | NA | NA | NA | 169.9 | 26.77 | NA | 1.08 | 3.25 | 5.09 | |||||||||||||||
| Range | 0.69–2.05 | NA | NA | NA | NA | 128.0–251.2 | 13.22–37.11 | NA | 0.33–4.0 | 1.86–4.63 4.23–7.86 | ||||||||||||||||
PO, per os; IV, intravenously; F, bioavailability; Vd, volume of distribution; Cl, clearance; AUC, area under the curve; C, maximum concentration; Cmin, minimum concentration; T,
maxmax
time to maximum concentration; T½, elimination half-life; MRT, mean residence time; SD, standard deviation
Levetiracetam 429
with status epilepticus and acute repetitive seizures (cluster seizures) (Hardy et al., 2012) reported pharmacokinetic parameters similar to the previous two studies in healthy dogs (Dewey et al., 2008; Patterson et al., 2008), except for the shorter half-life (2.2–2.3 h) in clinical patients, compared with 3–4 h in healthy dogs. Long-term PB administration in the epileptic dogs may have resulted in the shorter LEV half-life (Moore et al., 2011; Hardy et al., 2012). In the clinical study, LEV concentrations were dose proportional and remained within or exceeded the proposed human therapeutic range 5–46 mg/ml (Folland and Moriarty, 2002; Juenke et al., 2002; Leppik et al., 2002; Johannessen et al., 2003) at all time-points. In dogs that received the 30 mg/kg LEV IV dose, the mean ± standard deviation plasma LEV concentrations were 57.0 ± 17.8,
39.0 ± 14.1 and 21.4 ± 5.0 mg/ml at 15, 45 and 180 min, respectively. For the dogs that received the 60 mg/kg LEV IV dose, the mean plasma LEV concentrations were 141.4 ± 56.3,
118.6 ± 61.6 and 61.7 ± 68.0 mg/ml at 15, 45 and 180 min, respectively (Hardy et al., 2012).
LEV disposition in cats is similar to that in dogs (Table 16.2) (Bailey et al., 2008; Carnes et al., 2011).
Pharmacokinetic Interactions and Adverse Reactions
LEV has a low potential for clinically relevant pharmacokinetic interactions with other medications due to negligible hepatic metabolism and protein binding, and lack of induction or inhibition of drug-metabolizing hepatic enzymes (Isoherranen et al., 2001; Benedetti et al., 2004; Patsalos, 2004). LEV does not affect the steady-state serum concentration of several AEMs including PB, diazepam, clonazepam and gabapentin in humans (Gidal et al., 2005), and PB and Br in dogs (Muñana et al., 2012). Concomitant administration of AEMs that induce cytochrome P450 metabolism, such as PB, carbamazepine and phenytoin, can increase LEV clearance in people resulting in significantly lower plasma LEV concentrations; however, the clinical importance of this effect is uncertain (Contin et al., 2004).
In healthy dogs, PB administration (at 2.0–3.3 mg/kg twice daily for 21 days) significantly altered the pharmacokinetics of LEV (Moore et al., 2011). Compared with values determined when LEV was administered alone, concurrent administration of PB resulted in a significant decrease in LEV peak concentration (Cmax) from 32.39 ± 6.76 to 18.22 ± 8.97, a decrease in elimination half-life (T1/2) from 3.43 ± 0.47 to 1.73 ± 0.22, and an increase in oral clearance from 124.93 ± 26.93 to 252.99 ± 135.43 ml/h/kg. The extent of these pharmacokinetic changes varied among individual dogs. It has been speculated that increased metabolism and clearance of LEV is due to induction of oxidative enzymes by PB. LEV oral dosage may need to be adjusted when concurrently administered with PB (Moore et al., 2011).
Studies in animal models of epilepsy have shown pharmacodynamic interactions between LEV and other AEMs. Pharmacodynamic interactions lead to changes in the effects of either medication without affecting their plasma concentrations. Combinations of LEV with other AEMs, particularly those enhancing GABA-ergic inhibition, lead to synergistic effects on seizure protection, which are not associated with more pronounced side-effects and pharmacokinetic interactions (Matagne, 2001; Klitgaard and Matagne, 2002; Dudra-Jastrzebska et al., 2009; Kaminski et al., 2009). In audiogenic seizuring mice, the combination of LEV with valproic acid, clonazepam, diazepam or PB produced an increase in anticonvulsant potency by a factor of 28, 23, 19 and 16, respectively (Matagne, 2001; Kaminski et al., 2009). In a similar experimental model, synergistic interactions of LEV with diazepam, gabapentin, felbamate and topiramate were identified (Donato Di Paola et al., 2007). In amygdala-kindled rats, co-administration of a low dose of LEV with valproic acid, clonazepam, PB or carbamazepine produced an increase in anticonvulsant potency by a factor of 3, 4, 2 and 2.5, respectively (Klitgaard and Matagne, 2002). This increase was even more significant when a higher dose of LEV was used in combination with the other AEMs. In the murine pentylenetetrazole-induced seizure model, co-administration of LEV with clonazepam, ethosuximide, phenobarbital and valproic acid
Levetiracetam 431
exerted a supra-additive (synergistic) effect in suppressing seizures (Dudra-Jastrzebska et al., 2009). In the rat model of self-sustained status epilepticus, LEV was more efficacious than diazepam and potentiated diazepam’s anticonvulsant effects when used in combination (Mazarati et al., 2004). In the murine maximal electroshock-induced seizure model co-administration of LEV and felbamate resulted in pharmacodynamic supra-additivity as well as a pharmacokinetic interaction characterized by an increase in brain LEV concentrations (Luszczki et al., 2007). In addition, there are anecdotal clinical reports and experimental evidence of negative pharmacodynamic interactions of LEV. Combination of LEV with topiramate and carbamazepine could result in increased symptomatic neurotoxicity of topiramate and carbamazepine (Sisodiya et al., 2002; Luszczki et al., 2005).
The most commonly observed adverse effects associated with LEV administration in adults include somnolence, asthenia (generalized weakness), dizziness and upper respiratory infection (Cereghino etal.,2000; Abou-Khalil, 2008; Mbizvo et al., 2012). Non-specific changes in behaviour including hostility and nervousness have been reported as the most common LEV-related adverse effects in children (Verrotti et al., 2010; Mbizvo et al., 2012). Reported adverse effects attributed to LEV in dogs and cats appear infrequent and include sedation, ataxia, decreased appetite and vomiting in dogs (Volk et al., 2008; Muñana et al., 2012) and transient mild lethargy and inappetence in cats (Bailey et al., 2008). Dose reduction or permanent discontinuation has been necessary in a few cases to resolve these adverse effects. Transient mild to moderate hypersalivation has been reported in cats following administration of the commercially available LEV oral suspension (Carnes et al., 2011). The potential for toxicosis associated with LEV administration is low. In dogs, oral administration of 2000 mg/kg and IV administration of 1200 mg/kg resulted in mild adverse reactions, including salivation, vomiting, tachycardia and restlessness (UCB Pharma Inc, Smyrna, Georgia: unpublished data, 2001).
LEV administration does not appear to result in any clinically relevant effects on routine laboratory parameters (including haematology, serum chemistry profile or urinalysis) in dogs and cats (Carnes et al., 2011; Muñana et al., 2012), nor to affect serum PB and Br concentrations in dogs (Muñana et al., 2012).
Dosing and Monitoring Recommendations
Based on results of pharmacokinetic studies (Isoherranen et al., 2001; Moore et al., 2010; Carnes et al., 2011) and on the reference ranges suggested in humans (Folland and Moriarty, 2002; Juenke et al., 2002; Leppik et al., 2002; Johannessen et al., 2003) the recommended maintenance dosage of LEV for dogs and cats is 20 mg/kg, orally, every 8 h. The same dosage can be administered parenterally (SC, IM, IV in dogs; IV in cats) when oral administration is not possible (Patterson et al., 2008; Carnes et al., 2011; Hardy et al., 2011). Currently there are no studies on SC and IM administration in cats. One author has suggested increasing the daily dose by 20 mg/kg increments until efficacy is achieved, adverse effects become apparent, or the medication becomes cost-prohibitive (Dewey, 2006). In some cats, dosing every 6 h or a dosage of 40 mg/kg LEV every 8 h may be required to maintain serum LEV concentrations in the upper part of the reference range suggested in humans (Carnes et al., 2011).
A dosage of 30 mg/kg q12 h of XR LEV administered as Keppra XR® has been suggested (Beasley and Boothe, 2012).
LEV dosage reduction should be considered in patients with impaired renal function as this could result in decreased LEV renal clearance.
The LEV oral dose may need to be increased or dosing interval may need to be decreased when concurrently administered with PB, as PB has been shown to affect disposition of LEV in dogs (Moore et al., 2011).
Frequency of oral daily dosing may be reduced to once or twice daily with the XR LEV formulation. A dose of 30 mg/kg of XR LEV administered every 12–24 h may be adequate in dogs, although monitoring serum concentrations is recommended to individualize dosing (Beasley and Boothe, 2012). XR LEV could result in improved compliance and relatively constant plasma concentrations (Platt, 2011). However, further studies are currently needed to provide specific dosing recommendations with XR LEV.
When suggested reference serum LEV concentrations need to be reached rapidly, a single dose of 60 mg/kg can be administered orally or parenterally as loading and followed after 8 h by the maintenance dosage. Multiple administrations of 20 mg/kg of LEV can be used to treat acute repetitive seizures (ARS) (cluster seizures) and status epilepticus (SE) (see Chapters 23 and 24, this volume) any time hepatic dysfunction is suspected and therefore hepatically metabolized medications (e.g. benzodiazepines, PB) should be avoided or in animals resistant to these medications.
A reference range for LEV has not been definitively established in humans and suggested ranges for seizure control have been reported as 12–46 mg/ml (Leppik et al., 2002), 13–42 mg/ml (Folland and Moriarty, 2002), 5–30 mg/ml (Juenke et al., 2002) and 5–45 mg/ ml (Johannessen et al., 2003). There is no information available regarding a reference range in dogs and cats and therefore the reported human range is currently used as guideline. The main benefit of serum LEV concentration monitoring is to help to individualize treatment. If a baseline serum concentration is obtained during a period of satisfactory seizure control, the serum concentration can be re-evaluated in case of breakthrough seizures to assess if a drop in concentration played a role. Sampling time in relation to dose ingestion is important for the interpretation of serum LEV concentration due to LEV’s relatively short half-life. Collection of a trough sample allows assessment of the lowest concentration in the dosing interval as well as consistency of time of sampling. Additionally, time of the day (e.g. morning versus afternoon) should be consistent as a diurnal variation in LEV excretion may occur (Moore et al., 2010). Following oral administration of LEV at approximately 20 mg/kg every 8 h in healthy dogs, trough plasma LEV concentrations were 18.42 ± 5.16 mg/ml in the morning and 12.57 ± 4.34 mg/ml at midday, respectively (Moore et al., 2010). Assessment of steady-state serum or plasma LEV peak and trough concentrations can be used to determine LEV half-life. This can be particularly useful in dogs that are concurrently administered PB in order to identify the need to increase either total dosage administered or dosing interval of LEV (Moore et al., 2011).
In performing therapeutic monitoring of LEV, it has been recommended to separate serum or plasma from whole blood rapidly, to avoid LEV hydrolysis in the blood collection tube that would result in spuriously lower concentrations being measured (Patsalos et al., 2006).
In a recent study in epileptic dogs resistant to PB and Br, no association was identified between serum LEV concentrations and response to treatment (Muñana et al., 2012).
Efficacy
Double-blinded randomized clinical trials in humans have demonstrated that LEV is efficacious as adjunctive therapy for refractory focal-onset seizures, primary generalized tonic-clonic seizures, and myoclonic seizures of juvenile myoclonic epilepsy. In addition, LEV efficacy was equivalent to controlled release carbamazepine as first-line therapy for focal-onset seizures (Abou-Khalil, 2008).
Data on clinical efficacy of LEV in dogs are limited (Steinberg and Faissler, 2004; Bailey et al., 2008; Volk et al., 2008; Muñana et al., 2012). Two open-label studies on LEV as adjunctive therapy for refractory epilepsy in dogs reported a favourable treatment response during the initial 2–3 months compared to baseline (Steinberg and Faissler, 2004; Volk et al., 2008), however, a randomized, blinded trial did not demonstrate LEV efficacy compared to placebo. The randomized blinded trial included 34 idiopathic epileptic dogs resistant to PB and Br. Dogs received LEV (20 mg/kg PO q8h) or placebo for 16 weeks, and after a 4-week washout were crossed over to the alternate treatment for 16 weeks. Twenty-two (65%) dogs completed the study. Weekly seizure frequency decreased significantly during LEV administration relative to baseline (1.9 ± 1.9 to 1.1 ± 1.3 seizures/week),
Levetiracetam 433
however this reduction was not significant when compared to placebo. Of the 28 dogs completing the first treatment period (16 weeks), seizure freedom was reached in 17% of dogs on LEV and 0% of dogs on placebo and a ³50% reduction in seizure frequency was achieved in 56% of dogs on LEV and 30% of dogs on placebo, although these differences between LEV and placebo groups were not significant. Owner-perceived quality of life was significantly improved during LEV administration compared to placebo (Muñana et al., 2012). More favourable results may be achieved with higher LEV doses, a larger number of dogs completing the study, LEV used as first line AEM as well as in dogs with less severe forms of epilepsy.
The first open-label study included 15 idiopathic epileptic dogs with generalized seizures resistant to PB and/or Br. Adjunctive LEV at 7.1–23.8 mg/kg three times a day resulted in an overall reduction in seizure frequency (during the initial 3 months) of 54% compared to baseline (Steinberg and Faissler, 2004). In the second open-label study, 8 of 14 idiopathic epileptic dogs (57%) resistant to PB and Br experienced a reduction in seizure frequency ³50% following administration of LEV at 10 mg/kg three times a day for the initial 2 months. One additional dog responded when LEV dosage was increased to 20 mg/kg three times a day. LEV responders had a significant decrease in seizure frequency of 77%
(7.9 ± 5.2 to 1.8 ± 1.7 seizures/month) and a decrease in seizure days per month of 68%
(3.8 ± 1.7 to 1.2 ± 1.1 seizure days/month). However, six out of nine (67%) responders experienced an increase in seizure frequency and seizure days per month 4–8 months after initiation of LEV (Volk et al., 2008). This increase in seizure frequency may have been due to progression of the epilepsy, natural fluctuations of seizure frequency, poor owner compliance or pharmacotolerance (reduction in response to a medication after repeated administration). Development of tolerance to LEV has been reported in one animal model of epilepsy (Löscher and Hönack, 2000), however it does not seem to be a major problem in humans. Several studies on long-term efficacy of LEV in epileptic humans have shown that response appears to be maintained for the majority of patients, but a small percentage of individuals may have a reduction in benefit while others may have an improvement (Abou-Khalil, 2008).
The concerns about development of tolerance and the cost of LEV have prompted the use of LEV as intermittent pulse therapy in epileptic dogs that tend to have seizures (particularly as clusters or status epilepticus) at predictable time intervals (e.g. every fourth week). In these dogs LEV can be started just before the time of the expected seizures or soon after the first seizure and continued for a couple of days. A similar approach can be used when the pet owner recognizes the prodrome so that the LEV can be administered at this stage at 20–60 mg/kg once and continued at 20 mg/kg every 8 h for a couple of days. There is no published study at present evaluating the efficacy of this type of LEV intermittent pulse treatment but the authors’ initial anecdotal impressions are positive.
In the human literature, observational studies have supported a role for oral and intravenous LEV in the treatment of acute repetitive seizures (cluster seizures) and status epilepticus (SE) that were refractory to initial standard therapy (Aiguabella et al, 2011; McTague et al., 2012). A randomized, placebo-controlled, double-masked study including 19 dogs with SE or cluster seizures (acute repetitive seizures) has shown that administration of intravenous LEV (30 or 60 mg/kg) in addition to diazepam resulted in a significantly higher responder rate (56% after LEV) compared to placebo and diazepam (10%). In addition, dogs in the placebo group required significantly more boluses of diazepam compared with dogs in the LEV group (Hardy et al., 2012). Further studies are required to investigate LEV efficacy in SE and cluster seizures in a larger number of dogs and to determine the maximum safe dosage of IV LEV, efficacy of single versus multiple doses of LEV, the optimal dosing interval of LEV, and timing of LEV treatment relative to administration of benzodiazepines or other AEMs.
LEV may also be efficacious in preventing post-operative seizures in dogs undergoing portosystemic shunt surgery. A retrospective study including 126 dogs concluded that administration of LEV at 20 mg/kg orally q8h for a minimum of 24 h before surgery significantly decreased the risk of post-operative seizures and death in dogs undergoing surgical attenuation of extrahepatic congenital portosystemic shunts with ameroid ring constrictors (Fryer et al., 2011).
Information on LEV efficacy in cats is limited to one open-label, non-comparative study, including 12 epileptic cats considered resistant to PB (Bailey et al., 2008). Administration of LEV at 20 mg/kg orally q8h resulted in a significant decrease in median seizure frequency (from 2.1 seizures/month before initiation of LEV to 0.42 seizures/month after initiation of LEV). Seven of ten cats in which seizure logs were available had a reduction in seizure frequency ³50% with a median percentage reduction in seizure frequency of 92%. The reduction in seizure frequency ³50% was maintained in five of seven cats with a follow-up ³8 months (Bailey, 2008). There is also an anecdotal report of LEV efficacy as mono-therapy in epileptic cats and as adjunctive treatment to PB in cats undergoing surgical resection of intracranial meningiomas (Bailey and Dewey, 2009).
Summary Recommendations
• LEV is well tolerated and generally safe for use in the dog and cat.
References
Abou-Khalil, B. (2008) Levetiracetam in the treatment of epilepsy. Neuropsychiatric Disease and Treatment 4(3), 507–523.
Aiguabella, M., Falip, M., Villanueva, V., de la Peña, P., Molins, A., Garcia-Morales, I., Saiz, R.A., Pardo, J., Tortosa, D., Sansa, G. and Miró, J. (2011) Efficacy of intravenous levetiracetam as an add-on treatment in status epilepticus: a multicentric observational study. Seizure 20, 60–64.
Archer, D.P., Lamberty, Y., Wang, B., Davis, M.J., Samanani, N. and Roth, S.H. (2007) Levetiracetam reduces anesthetic-induced hyperalgesia in rats. Anesthesia and Analgesia 104, 180–185.
Ardid, D., Lamberty, Y., Alloui, A., Coudore-Civiale, M.A., Klitgaard, H. and Eschalier, A. (2003) Antihyperalgesic effect of levetiracetam in neuropathic pain models in rats. European Journal of Pharmacology 473, 27–33.
Bailey, K.S. and Dewey, C.W. (2009) The seizuring cat: diagnostic work-up and therapy. Journal of Feline Medicine and Surgery 11, 385–394.
Bailey, K.S., Dewey, C.W., Boothe, D.M., Barone, G. and Kortz, G.D. (2008) Levetiracetam as an adjunct to Phenobarbital treatment in cats with suspected idiopathic epilepsy. Journal of the American Veterinary Medical Association 232, 867–872.
Levetiracetam 435
Beasley, M.J. and Boothe, D.M. (2012) The pharmacokinetics of single dose extended release Keppra® with and without food in healthy adult dogs. ACVIM Forum Abstract Proceeding, p. 819.
Belcastro, V., Pierguidi, L. and Tambasco, N. (2011) Levetiracetam in brain ischemia: clinical implications in neuroprotection and prevention of post-stroke epilepsy. Brain and Development 33(4), 289–293.
Benedetti, M.S., Coupez, R., Whomsley, R., Nicolas, J.M., Collart, P. and Baltes, E. (2004) Comparative pharmacokinetics and metabolism of levetiracetam, a new anti-epileptic agent, in mouse, rat, rabbit and dog. Xenobiotica 34, 281–300.
Berkovic, S.F., Knowlton, R.C., Leroy, R.F., Schiemann, J. and Falter, U. (2007) Placebo-controlled study of levetiracetam in idiopathic generalized epilepsy. Neurology 69, 1751–1760.
Carnes, M.B., Axlund, T.W. and Boothe, D.M. (2011) Pharmacokinetics of levetiracetam after oral and intravenous administration of a single dose to clinically normal cats. American Journal of Veterinary Research 72, 1247–1252.
Cereghino, J.J., Biton, V., Abou-Khalil, B., Dreifuss, F., Gauer, L.J. and Leppik I. (2000) Levetiracetam for partial seizures: results of a double-blind, randomized clinical trial. Neurology 55, 236–242.
Christensen, K.V., Leffers, H., Watson, W.P., Sánchez, C., Kallunki, P. and Egebjerg, J. (2010) Levetiracetam attenuates hippocampal expression of synaptic plasticity-related immediate early and late response genes in amygdala-kindled rats. BMC Neuroscience 11, 9.
Contin, M., Albani, F., Riva, R. and Baruzzi, A. (2004) Levetiracetam therapeutic monitoring in patients with epilepsy: Effect of concomitant antiepileptic drugs. Therapeutic Drug Monitoring 26, 375–379.
De Deyn, P.P., Kabatu, H., D’Hooge, R. and Mori, A. (1992) Protective effect of ucb L059 against postural stimulation-induced seizures in EL Mice. Neuroscience 18, 187–192.
De Smedt, T., Vonck, K., Raedt, R., Dedeurwaerdere, S., Claeys, P., Legros, B., Wyckhuys, T., Wadman, W. and Boon, P. (2005) Rapid kindling in preclinical anti-epileptic drug development: the effect of levetiracetam. Epilepsy Research 67(3), 109–116.
Dewey, C.W. (2006) Anticonvulsant therapy in dogs and cats. Veterinary Clinics of North America – Small Animal Practice 36(5), 1107–1127.
Dewey, C.W., Barone, G., Boothe, D.M., Smith, K. and O’Connor J.H. (2005) The use of oral levetiracetam as an add-on anticonvulsant drug in cats receiving phenobarbital [abstract]. Journal of Veterinary Internal Medicine 19, 458.
Dewey, C.W., Bailey, K.S., Boothe, D.M., Badgley, B.L. and Cruz-Espindola, C. (2008) Pharmacokinetics of single-dose intravenous levetiracetam administration in normal dogs. Journal of Veterinary Emergency and Critical Care 18, 153–157.
Doheny, H.C., Ratnaraj, N., Whittington, M.A., Jefferys, J.G. and Patsalos, P.N. (1999) Blood and cerebrospinal fluid pharmacokinetics of the novel anticonvulsant levetiracetam (ucb L059) in the rat. Epilepsy Research 34, 161–168.
Donato Di Paola, E., Gareri, P., Davoli, A., Gratteri, S., Scicchitano, F., Naccari, C. and De Sarro, G. (2007) Influence of levetiracetam on the anticonvulsant efficacy of conventional antiepileptic drugs against audiogenic seizures in DBA/2 mice. Epilepsy Research 75, 112–121.
Dudra-Jastrzebska, M., Andres-Mach, M.M., Ratnaraj, N., Patsalos, P.N., Czuczwar, S.J. and Luszczki, J.J. (2009) Isobolographic characterization of the anticonvulsant interaction profiles of levetiracetam in combination with clonazepam, ethosuximide, phenobarbital and valproate in the mouse pentylenetetrazole-induced seizure model. Seizure 18(9), 607–614.
Fang, Y., Wu, X., Xu, L., Tang, X., Wang, J., Zhu, G., Hong, Z. (2014) Randomized-controlled trials of levetiracetam as an adjunctive therapy in epilepsy of multiple seizure types. Journal of Clinical Neuroscience 21, 55–62.
Folland, C. and Moriarty, G.L. (2002) Clinical experience of levetiracetam (LEV) in refractory adult epilepsy patients. Epilepsia 43(Suppl. 7), 192.
Fryer, K.J., Levine, J.M., Peycke, L.E., Thompson, J.A. and Cohen, N.D. (2011) Incidence of postoperative seizures with and without levetiracetam pretreatment in dogs undergoing portosystemic shunt attenuation. Journal of Veterinary Internal Medicine 25, 1379–1384.
Gidal, B.E., Baltes, E., Otoul, C. and Perucca, E. (2005) Effect of levetiracetam on the pharmacokinetics of adjunctive antiepileptic drugs: A pooled analysis of data from randomized clinical trials. Epilepsy Research 64, 1–11.
Gower, A.J., Hirsch, E., Boehrer, A., Noyer, M. and Marescaux, C. (1995) Effects of levetiracetam, a novel antiepileptic drug, on convulsant activity in two genetic rat models of epilepsy. Epilepsy Research 22(3), 207–213.
Hanon, E. and Klitgaard, H. (2001) Neuroprotective properties of the novel antiepileptic drug levetiracetam in the rat middle cerebral artery occlusion model of focal cerebral ischemia. Seizure 10, 287–293.
Hardy, B.T., Patterson, E. and Cloyd, J.M. (2011) Subcutaneous administration of levetiracetam in healthy dogs. ACVIM Forum Abstract Proceeding, p. 741.
Hardy, B.T., Patterson, E.E., Cloyd, J.M., Hardy, R.M. and Leppik, I.E. (2012) Double-masked, placebo-controlled study of intravenous levetiracetam for the treatment of status epilepticus and acute repetitive seizures in dogs. Journal of Veterinary Internal Medicine 26, 334–340.
Isoherranen, N., Yagen, B., Soback, S., Roeder, M., Schurig, V. and Bialer, M. (2001) Pharmacokinetics of levetiracetam and its enantiomer (R)-a-ethyl-2-oxo-pyrrolidine acetamide in dogs. Epilepsia 42, 825–830.
Johannessen, S.I., Battino, D., Berry, D.J., Bialer, M., Kramer, G., Tomson, T. and Patsalos, P.N. (2003) Therapeutic drug monitoring of the newer antiepileptic drugs. Therapeutic Drug Monitoring 25, 347–363.
Juenke, J.M., Brown, P.I. and Urry, F.M. (2002) Analysis of levetiracetam and zonisamide in plasma and serum by HPLC. Clinical Chemistry 48(Suppl. 6), 44–45.
Kaminski, R.M., Matagne, A., Patsalos, P.N. and Klitgaard, H. (2009) Benefit of combination therapy in epilepsy: a review of the preclinical evidence with levetiracetam. Epilepsia 50(3), 387–397.
Klitgaard, H. and Matagne, A. (2002) Levetiracetam enhances markedly the seizure suppression of other antiepileptic drugs in amygdala-kindled rats. Epilepsia 43, 219.
Klitgaard, H., Matagne, A., Gobert, J. and Wülfert, E. (1998) Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. European Journal of Pharmacology 24, 191–206.
Komur, M., Okuyaz, C., Celik, Y., Resitoglu, B., Polat, A., Balci, S., Tamer, L., Erdogan, S., Beydagi, H. (2014) Neuroprotective effect of levetiracetam on hypoxic ischemic brain injury in neonatal rats. Child's Nervous System 30, 1001–1009.
Krauss, G.L., Betts, T., Abou-Khalil, B., Gergey, G., Yarrow, H. and Miller, A. (2003) Levetiracetam treatment of idiopathic generalised epilepsy. Seizure 12, 617–620.
Lee, C.Y., Chen, C.C. and Liou, H.H. (2009) Levetiracetam inhibits glutamate transmission through presynaptic P/Q-type calcium channels on the granule cells of the dentate gyrus. British Journal of Pharmacology 158(7), 1753–1762.
Leppik, I.E., Rarick, J.O., Walezak, T.S., Tran, T.A., White, J.R. and Gumnit, R.J. (2002) Effective levetiracetam doses and serum concentrations: Age effects. Epilepsia 43(Suppl. 7), 240.
Lo, B.W., Kyu, H.H., Jichici, D., Upton, A.M., Akl, E.A. and Meade, M.O. (2011) Meta-analysis of randomized trials on first line and adjunctive levetiracetam. The Canadian Journal of Neurological Science 38(3), 475–486.
Löscher, W. and Hönack, D. (1993) Profile of ucb L059, a novel anticonvulsant drug, in models of partial and generalized epilepsy in mice and rats. European Journal of Pharmacology 232(2–3), 147–158.
Löscher, W. and Hönack, D. (2000) Development of tolerance during chronic treatment of kindled rats with the novel antiepileptic drug levetiracetam. Epilepsia 41, 1499–1506.
Löscher, W., Hönack, D. and Rundfeldt, C. (1998) Antiepileptogenic effects of the novel anticonvulsant levetiracetam UCB (L059) in the kindling model of temporal lobe epilepsy. The Journal of Pharmacology and Experimental Therapeutics 284, 474–479.
Leniger, T., Thone, J., Bonnet, U., Hufnagel, A., Bingmann, D. and Wiemann, M. (2004) Levetiracetam inhibits Na+-dependent Cl-/HCO3- exchange of adult hippocampal CA3 neurons from guinea-pigs. British Journal of Pharmacology 142, 1073–1080.
Lukyanetz, E.A., Shkryl, V.M. and Kostyuk, P.G. (2002) Selective blockade of N-type calcium channels by levetiracetam. Epilepsia 43, 9–18.
Luszczki, J.J., Andres, M.M., Czuczwar, P., Cioczek-Czuczwar, A., Wojcik-Cwikla, J., Ratnaraj, N., Patsalos, P.N. and Czuczwar, S.J. (2005) Levetiracetam selectively potentiates the acute neurotoxic effects of topiramate and carbamazepine in the rotarod test in mice. European Neuropsychopharmacology 15(6), 609–616.
Luszczki, J.J., Andres-Mach, M.M., Ratnaraj, N., Patsalos, P.N. and Czuczwar, S.J. (2007) Levetiracetam and felbamate interact both pharmacodynamically and pharmacokinetically: an isobolographic analysis in the mouse maximal electroshock model. Epilepsia 48(4), 806–815.
Lynch, B.A., Lambeng, N., Nocka, K., Kensel-Hammes, P., Bajjalieh, S.M., Matagne, A. and Fuks, B. (2004) The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proceedings of the National Academy of Sciences of the United States of America 101, 9861–9866.
Lyseng-Williamson, K.A. (2011) Spotlight on levetiracetam in epilepsy. CNS Drugs 25, 901–905.
Madeja, M., Margineanu, D.G., Gorji, A., Siep, E., Boerrigter, P., Klitgaard, H. and Speckmann, E.J. (2003) Reduction of voltage-operated potassium currents by levetiracetam: a novel antiepileptic mechanism of action? Neuropharmacology 45, 661–671.
Levetiracetam 437
Margineanu, D.G. and Klitgaard, H. (2000) Inhibition of neuronal hypersynchrony in vitro differentiates levetiracetam from classical antiepileptic drugs. Pharmacology Research 42, 281–285.
Margineanu, D.G. and Klitgaard, H. (2003) Levetiracetam has no significant gamma-aminobutyric acid-related effect on paired-pulse interaction in the dentate gyrus of rats. European Journal of Pharmacology 18, 466(3), 255–261.
Matagne, A. (2001) Levetiracetam enhances markedly the seizure suppression of other antiepileptic drugs in audiogenic susceptible mice. Epilepsia 42, 82.
Mazarati, A.M., Baldwin, R., Klitgaard, H., Matagne, A. and Wasterlain, C.G. (2004) Anticonvulsant effects of levetiracetam and levetiracetam-diazepam combinations in experimental status epilepticus. Epilepsy Research 58(2–3), 167–174.
Mbizvo, G.K., Dixon, P., Hutton, J.L. and Marson, A.G. (2012) Levetiracetam add-on for drug-resistant focal epilepsy: an updated Cochrane Review. Cochrane Database Systematic Reviews Sep 12; 9: CD001901.
McTague, A., Kneen, R., Kumar, R., Spinty, S. and Appleton, R. (2012) Intravenous levetiracetam in acute repetitive seizures and status epilepticus in children: experience from a children’s hospital. Seizure 21(7), 529–534.
Micov, A., Tomic´, M., Popovic´, B. and Stepanovic´-Petrovic´, R. (2010) The antihyperalgesic effect of levetiracetam in an inflammatory model of pain in rats: mechanism of action. British Journal of Pharmacology 161, 384–392.
Moore, S.A., Muñana, K.R., Papich, M.G. and Nettifee-Osborne, J. (2010) Levetiracetam pharmacokinetics in healthy dogs following oral administration of single and multiple doses. American Journal of Veterinary Research 71, 337–341.
Moore, S.A., Muñana, K.R., Papich, M.G. and Nettifee-Osborne J.A. (2011) The pharmacokinetics of levetiracetam in healthy dogs concurrently receiving phenobarbital. Journal of Veterinary Pharmacology and Therapeutics 34, 31–34.
Muñana, K.R., Thomas, W.B., Inzana, K.D., Nettifee-Osborne, J.A., McLucas, K.J., Olby, N.J., Mariani, C.J. and Early, P.J. (2012) Evaluation of levetiracetam as adjunctive treatment for refractory canine epilepsy: a randomized, placebo-controlled, crossover trial. Journal of Veterinary Internal Medicine 26, 341–348.
Niespodziany, I., Klitgaard, H. and Margineanu, D.G. (2001) Levetiracetam inhibits the high-voltage-activated Ca(2+) current in pyramidal neurones of rat hippocampal slices. Neuroscience Letters 306(1–2), 5–8.
Niespodziany, I., Klitgaard, H. and Margineanu, D.G. (2003) Desynchronizing effect of levetiracetam on epileptiform responses in rat hippocampal slices. Neuroreport 14, 1273–12176.
Noachtar, S., Andermann, E., Meyvisch, P., Andermann, F., Gough, W.B. and Schiemann-Delgado, J. (2008) Levetiracetam for the treatment of idiopathic generalized epilepsy with myoclonic seizures. Neurology 70, 607–616.
Otoul, C., Arrigo, C., Van Rijckevorsel, K. and French, J.A. (2005) Meta-analysis and indirect comparisons of levetiracetam with other second-generation antiepileptic drugs in partial epilepsy. Clinical Neuropharmacology Mar–Apr, 28(2), 72–78.
Ozcan, M., Ayar, A., Canpolat, S. and Kutlu, S. (2008) Antinociceptive efficacy of levetiracetam in a mice model for painful diabetic neuropathy. Acta Anaesthesiologica Scandinava 52, 926–930.
Patsalos, P.N. (2004) Clinical pharmacokinetics of levetiracetam. Clinical Pharmacokinetics 43(11), 707–724.
Patsalos, P.N., Ghattaura, S., Ratnaraj, N. and Sander, J.W. (2006) In situ metabolism of levetiracetam in blood of patients with epilepsy. Epilepsia 47, 1818–1821.
Patterson, E.E., Goel, V., Cloyd, J.C., O’Brien, T.D., Fisher, J.E., Dunn, A.W. and Leppik I.E. (2008) Intramuscular, intravenous and oral levetiracetam in dogs: safety and pharmacokinetics. Journal of Veterinary Pharmacology and Therapeutics 31, 253–258.
Peters, R.K., Schubert, T., Clemmons, R. and Vickroy, T. (2014) Levetiracetam Rectal Administration in Healthy Dogs. Journal of Veterinary Internal Medicine. Jan 13. doi: 10.1111/jvim.12269.
Pisani, A., Bonsi, P., Martella, G., De Persis, C., Costa, C., Pisani, F., Bernardi, G. and Calabresi, P. (2004) Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia 45, 719–728.
Platt, S.R., Freeman, A.C., Aljuffali, I.A., Gullick, D. and Arnold, R. (2011) Pharmacokinetic evaluation of extended release levetiracetam in dogs. ACVIM Forum Abstract Proceeding, p. 729.
Rekling, J.C. (2003) Neuroprotective effects of anticonvulsants in rat hippocampal slice cultures exposed to oxygen/glucose deprivation. Neuroscience Letters 335, 167–170.
Rigo, J.M., Hans, G., Nguyen, L., Rocher, V., Belachew, S., Malgrange, B., Leprince, P., Moonen, G., Selak, I., Matagne,
A. and Klitgaard, H. (2002) The anti-epileptic drug levetiracetam reverses the inhibition by negative allosteric modulators of neuronal GABA- and glycine-gated currents. British Journal of Pharmacology 136, 659–672.
Rossetti, A.O., Jeckelmann, S., Novy, J., Roth, P., Weller, M., Stupp, R. (2014) Levetiracetam and pregabalin for antiepileptic monotherapy in patients with primary brain tumors. A phase II randomized study. Neuro-oncology 16(4), 584–588.
Shetty, A.K. (2013) Prospects of levetiracetam as a neuroprotective drug against status epilepticus, traumatic brain injury, and stroke. Frontiers in Neurology 4: 172.
Sisodiya, S.M., Sander, J.W. and Patsalos, P.N. (2002) Carbamazepine toxicity during combination therapy with levetiracetam: a pharmacodynamic interaction. Epilepsy Research 48(3), 217–219.
Sliva, J., Dolezal, T., Prochazkova, M., Votava, M. and Krsiak, M. (2008) Preemptive levetiracetam decreases postoperative pain in rats. Neuro Endocrinology Letters 29, 953–957.
Steinberg, M. and Faissler, D. (2004) Levetiracetam therapy for long-term idiopathic epileptic dogs [abstract]. Journal of Veterinary Internal Medicine 18, 410.
Stephen, L.J., Kelly, K., Parker, P. and Brodie, M.J. (2011) Levetiracetam monotherapy – outcomes from an epilepsy clinic. Seizure 20(7), 554–557.
Stienen, M.N., Haghikia, A., Dambach, H., Thöne, J., Wiemann, M., Gold, R., Chan, A., Dermietzel, R., Faustmann, P.M., Hinkerohe, D. and Prochnow, N. (2011) Anti-inflammatory effects of the anticonvulsant drug levetiracetam on electrophysiological properties of astroglia. British Journal of Pharmacology 162, 491–507.
Stratton, S.C., Large, C.H., Cox, B., Davies, G. and Hagan, R.M. (2003) Effects of lamotrigine and levetiracetam on seizure development in a rat amygdala kindling model. Epilepsy Research 53(1–2), 95–106.
Verrotti, A., D’Adamo, E., Parisi, P., Chiarelli, F. and Curatolo, P. (2010) Levetiracetam in childhood epilepsy. Paediatric Drugs 12(3), 177–186.
Volk, H.A., Matiasek, L.A., Luján Feliu-Pascual, A., Platt, S.R. and Chandler, K.E. (2008) The efficacy and tolerability of levetiracetam in pharmacoresistant epileptic dogs. Veterinary Journal 176, 310–319.
Yang, X.F., Weisenfeld, A. and Rothman, S.M. (2007) Prolonged exposure to levetiracetam reveals a pre-synaptic effect on neurotransmission. Epilepsia 48, 1861–1869.
Zona, C., Niespodziany, I., Marchetti, C., Klitgaard, H., Bernardi, G. and Margineanu, D.G. (2001) Levetiracetam does not modulate neuronal voltage-gated Na+ and T-type Ca2+ currents. Seizure 10(4), 279–286.
17 Gabapentin and Pregabalin
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Gabapentin (GBP) and pregabalin (PGB) are structurally related compounds (Fig. 17.1) with recognized anti-epileptic, antihyperalgesic and anti-allodynic efficacy in people and animal experimental models. PGB seems more potent and effective than GBP (Bockbrader et al., 2010; Delahoyetal., 2010). Both medications are believed to mediate their therapeutic action primarily through their high-affinity binding to alpha2delta auxiliary subunit of specific voltage gated calcium channels in neurons (Field et al., 2006).
Information on tolerability, safety and clinical efficacy of GBP and PGB is limited in dogs and cats (Vollmer et al., 1986; Radulovic et al., 1995; Govendir et al., 2005; Platt et al., 2006; Cashmore et al., 2009; Dewey et al., 2009; Salazar et al., 2009; Reid et al., 2010; Siao et al., 2010; Wagner et al., 2010; Wolfe and Poma, 2010; KuKanich and Cohen, 2011; Vettorato and Corletto, 2011; Aghighi et al., 2012; Lorenz et al., 2013; Steagall and Monteiro-Steagall, 2013).
The commercially available liquid formulation of gabapentin in the USA contains xylitol (300 mg/ml), which can be toxic, therefore a tablet or capsule formulation of gabapentin should be preferentially used.
Gabapentin
GBP, 1-(aminomethyl)cyclohexyl acetic acid, was originally developed as a chemical analogue
of g-aminobutyric acid (GABA) to attenuate polysynaptic spinal reflexes for the treatment of muscle spasticity (Fig. 17.1). Although the antispastic effects of GBP seemed to be modest, this medication was found to have anticonvulsant activity in a range of experimental seizure models (McLean, 1995). Subsequently GBP was also found to be efficacious in the treatment of neuropathic and post-operative pain (Rosner et al., 1996; Taylor et al., 1998; Bennett and Simpson, 2004; Coderre et al., 2005; Pandey et al., 2005; Cheng and Chiou, 2006; Clarke et al., 2012). GBP has been approved in Europe and by the US Food and Drug Administration (FDA) since 1993 for adjunctive treatment of focal seizures with or without secondary generalization and for treatment of post-herpetic neuralgia (Bockbrader, 2010). In 2011, the US FDA approved extended release GBP, which allows once daily dosing interval in people (Cundy et al., 2008; Gordi et al., 2008). GBP has been used in dogs as adjunctive anti-epileptic medication (AEMs) and for the treatment of neuropathic and post-operative pain (Govendir et al., 2005; Platt et al., 2006; Lamont, 2008; Cashmore et al., 2009; Wagner et al., 2010; Wolfe and Poma, 2010; Aghighi et al., 2012). Information on GBP in cats is limited to pharmacology studies (Reid et al., 2010; Siao et al., 2010) and a few case reports on treatment of chronic and peri-operative pain
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
(Vettorato and Corletto, 2011; Lorenz et al., 2013; Steagall and Monteiro-Steagall, 2013).
Mechanism of Action
GBP mechanism of action is incompletely understood. Although GBP is a structural analogue of the inhibitory neurotransmitter GABA, it does not bind to GABAA or GABAB receptors, it is not metabolized into GABA or a GABA agonist and it does not inhibit GABA uptake or degradation (Taylor et al., 1998; Jensen et al., 2002; Cheng and Chiou, 2006; Sills, 2006). GBP (as well as PGB) also differs from GABA because it readily crosses membrane barriers via L-amino acid transporters.
H2N COOH H2N
The predominant pharmacological mechanism of GBP (as well as PGB) is believed to be mediated by binding to the alpha2-delta-1 and alpha2-delta-2 subunits of voltage-gated calcium channels resulting in presynaptic inhibition of calcium influx, subsequent inhibition of excitatory neurotransmitter release and attenuation of postsynaptic excitability (Fig. 17.2) (Field et al., 2006). In addition, by binding to the alpha2-delta subunits, GBP has been shown to prevent formation of aberrant excitatory synapses, which may contribute to the pathophysiology of epilepsy and neuropathic pain (Eroglu et al., 2009). The selective presynaptic inhibitory effect on voltage-gated calcium channels containing alpha2-delta-1 subunit, may also account for some of the proposed additional effects of GBP, including
COOH H2N
![]() COOH
COOH
H H3C CH3
GABA Gabapentin Pregabalin

Fig. 17.2. Neuronal receptor targets for gabapentin and pregabalin.
activation of GABAB receptors (Bertrand et al., 2001, 2003; Parker et al., 2004), modulation of presynaptic NMDA receptors (Suarez et al., 2005; Kim et al., 2009) and the reduced release of both glutamate and other excitatory neurotransmitters (e.g. substance P, norepinephrine) (Maneuf et al., 2001, 2004; Cunningham et al., 2004; Sills, 2006). Other proposed cellular effects of GBP include modest actions on the GABA-ergic neurotransmitter system (Petroff et al., 1996) and on voltage-gated potassium channels (McClelland et al., 2004; Sills, 2006).
Metabolism and Pharmacokinetics
The metabolism and pharmacokinetics of GBP have been evaluated in several species (Vollmer et al., 1986; Radulovic et al., 1995; Siao et al., 2010; KuKanich and Cohen, 2011). GBP is well absorbed and peak plasma concentrations are reached 1–3 h following oral administration (Radulovic et al., 1995; Siao et al., 2010; KuKanich and Cohen, 2011). Food has no clinically relevant effect on the absorption of GBP, the principal site of which is the duodenum (Vollmer et al., 1986). Oral bioavailability was 80% in dogs administered 50 mg/kg (Radulovic et al., 1995) and 89% in cats administered 10 mg/kg, although interindividual variability was observed in cats (Siao et al., 2010). In dogs, area under the curve and maximum plasma GBP concentrations increase proportionally to the administered dose for doses ranging from 10 to 50 mg/kg (Radulovic et al., 1995; KuKanich and Cohen, 2011) (Table 17.1). In humans, GBP absorption relies on a saturable dose-dependent L-amino acid transport system in the small intestine resulting in dose-limited nonlinear absorption and inverse correlation between bioavailability and dose (Stewart et al., 1993; Berry et al., 2003). GBP bioavailability in humans varied from approximately 80% with an oral dose of 100 mg every 8 h to 27% with an oral dose of 1600 mg every 8 h (Stewart et al., 1993). The extended release GBP formulations have been developed to allow for dose-proportional absorption and bioavailability in humans (Cundy et al., 2008; Gordi et al., 2008). GBP readily crosses the blood-brain barrier, and at 1, 2, 4
Table 17.1. Pharmacokinetic parameters of GBP in healthy greyhounds administered a single oral dose of approximately 10 and 20 mg/kg, respectively (modified from KuKanich and Cohen, 2011) and in healthy beagle dogs administered a single oral or intravenous dose of GBP of 50 mg/kg (Radulovic et al., 1995).
Pharmacokinetic parameters
Dose AUCinf Relative Vz/F Cl/F (ml/ CTT½ kz MRTinf
max max
References administered (h·µg/m) F (l/kg) min/kg) (µg/ml) (h) (h) (h)
KuKanich 10 mg/kg PO Mean 48.77 1.13 0.983 3.49 8.54 1.31 3.25 5.43 and Cohen, Median 49.5 1.28 0.942 3.49 9.68 1.5 3.35 5.4 2011 Minimum 39.77 0.83 0.85 2.97 5.32 0.75 2.63 4.6 Maximum 63.02 1.41 1.256 4.26 10.9 2 3.68 6.78 20 mg/kg PO Mean 86.02 NA 1.17 3.96 13.22 1.51 3.41 5.69 Median 83.01 NA 1.207 3.97 12.95 1.75 3.39 5.4 Minimum 60 NA 0.835 2.65 10.7 1 3.07 5.01 Maximum 151.24 NA 1.476 5.56 18.2 2 3.91 6.94
Radulovic 50 mg/kg PO Mean 242 NA NA NA 56.3 1.1 2.2a NA et al., 1995 50 mg/kg IV Mean 367 NA 0.158 2.27 NA NA 2.9a NA
aHarmonic mean of half-life. PO, per os; IV, intravenously; AUCinf, area under the curve from time 0 to infinity; Relative F, relative fraction of the dose absorbed for each oral dose rate; Vz/F, apparent volume of distribution of the area fraction of the dose absorbed; Cl/F, plasma clearance per fraction of the dose absorbed; C, maximum plasma concentration; T, time
maxmax
to maximum plasma concentration; T½ kz, terminal half-life; MRTinf, mean residence time extrapolated to infinity; NA, not assessed
and 8 h post-dose, GBP concentrations in the canine brain are similar, although slightly lower to those in the blood (Vollmer et al., 1986). In dogs, approximately 30% to 40% of the orally administered dose of GBP is hepatically metabolized to N-methyl-gabapentin. This metabolite and the remainder of the parent drug are excreted primarily by the kidneys (Vollmer et al., 1986; Radulovic et al., 1995). In the mouse, rat, monkey and human, GBP undergoes minimal to no biotransformation; it is primarily excreted unchanged renally, and its elimination half-life ranges from 1.7 h in the rat to 6 h in humans (Vollmer et al., 1986; Radulovic et al., 1995). Plasma clearance is proportional to creatinine clearance. GBP binding to plasma proteins is <3% and hepatic microsomal CYP enzyme induction has not been observed (Radulovic et al., 1995).
Pharmacokinetic parameters of GBP in healthy dogs and cats are detailed in Tables 17.1 and 17.2, respectively (Radulovic et al., 1995; Siao et al., 2010; KuKanich and Cohen, 2011). Pharmacokinetics following multiple administrations of GBP in healthy beagle dogs (IV, 25 mg/kg/day for 14 days; or PO, 50 mg/ kg/day for 28 days) were comparable to those following single dose administration (Radulovic et al., 1995).
The pharmacokinetics of a sustained-release formulation (other than the ones currently approved for humans) of GBP were comparable to pharmacokinetics of the immediate-release formulation in healthy beagle dogs. However, in vitro the sustained-release tablet did not disintegrate for 12 h, whereas the immediate-release tablet disintegrated within 30 min and the time for 50% drug release (t50%) from the sustained-release tablet was over ten times slower than that from immediate-release tablet (Rhee et al., 2008).
The study on GPB pharmacokinetics in cats revealed high interindividual variability in bioavailability, Cmax and Tmax (Siao et al., 2010). These interindividual differences may have been due to the fact that administration of the GBP capsule was not followed by oral administration of water to flush the capsule through the oesophagus and that the cats had access to food ad libitum. These factors could have resulted in variability of GBP oesophageal and gastrointestinal transit time and absorption (Siao et al., 2010). However, high interindividual variability in serum GBP concentrations at any given dose has been detected also in humans (Gidal et al., 2000).
Based on simulations on a computer program, it was estimated that a dosage of GBP of 3 mg/kg PO every 6 h in cats would allow attainment and maintenance of plasma concentrations of 2 mg/ml, which have been associated with reduced seizure frequency in humans (Sivenius et al., 1991), and that administration of GBP at 8 mg/kg every 6 h in cats would result in plasma concentrations between 4.3 and 8 mg/ml, which appear to be associated with an analgesic effect in
Table 17.2. Pharmacokinetic parameters of GBP in healthy cats administered a single dose of GBP intravenously (4 mg/kg) or orally (10 mg/kg) (modified from Siao et al., 2010).
Pharmacokinetic parameters
Dose administered F (%) VdSS (l/kg) Cl (ml/min/kg) Cmax (µg/ml) Tmax (h) T1/2 γ (h)
4 mg/kg IV Mean ± SD NA 0.65 ± 0.014 3.02 ± 0.18 47.388 ± 5.102 NA 2.83 ± 0.34a Minimum NA 0.597 2.45 30.208 NA 2.52 Maximum 0.700 3.46 62.378 3.30
10 mg/kg PO Mean ± SD 88.7 ± 11.1 NA 2.99 ± 0.22 7.982 ± 1.053 1.67 ± 0.37 NA Minimum 49.6 NA 2.45 4.638 0.97 NA Maximum 118.3 3.46 10.550 2.92
aHarmonic mean ± jackknife pseudo-SD of T1/2 γ NA, not applicable; PO, per os; IV, intravenously; F, bioavailability; VdSS, apparent volume of distribution at steady state; Cl, clearance; C, maximum plasma concentration; T, time to reach the maximal plasma concentration; T1/2 γ ,
max max
elimination half-life.
people with neuropathic pain (Backonja and Glanzman, 2003; Siao et al., 2010).
Pharmacokinetic Interactions and Adverse Reactions
Although information in veterinary medicine is limited, pharmacokinetic interactions of GBP are unlikely to occur as GBP has negligible protein binding and does not induce hepatic CYP enzymes (Radulovic, 1995). Oral administration of GBP did not significantly alter the pharmacokinetic parameters of parental phenytoin in healthy beagle dogs (Matar et al., 2000). To date, no clinically relevant pharmacokinetic drug interactions have been identified in humans between GBP and other AEMs, with the exception of felbamate, whose half-life was prolonged by 46% and clearance was reduced by 37% (Hussein etal., 1996; Perucca, 2006; Johannessen Landmark and Patsalos, 2010). It was suggested that the reduced elimination of felbamate was due to active competition with GBP for renal excretion (Hussein et al., 1996). Co-administration of GBP and PB did not significantly affect mean trough steady-state PB concentrations as well as GBP C, T, AUC, T1/2 or urinary drug recovery
maxmax
compared to monotherapy (Hooper et al., 1991).
In humans, co-administration with antiacids containing aluminium or magnesium can decrease GBP absorption by up to 24% (LaRoche and Helmers, 2004; Johannessen Landmark and Patsalos, 2010). Co-administration with cimetidine (an inhibitor of renal tubular secretion) can reduce GBP renal clearance by approximately 14% (Radulovic et al., 1995). To date, no clinically significant pharmacodynamic interactions involving GBP have been reported (Johannessen Landmark and Patsalos, 2010).
The most commonly reported adverse effects of GBP in humans include dizziness, somnolence and fatigue (Marson et al., 2000; Bockbrader et al., 2010). These adverse effects are dose dependent and usually resolve within the first few weeks of treatment (LaRoche and Helmers, 2004; Bockbrader et al., 2010). Modest weight gains have also been observed, but no serious idiosyncratic reactions or organ toxicities have been identified in humans or experimental animals (LaRoche and Helmers, 2004). An increased frequency in adverse effects has been reported in humans with serum GBP concentrations greater than 25 mg/ml (146 mmol/l) (Johannessen et al., 2003).
Sedation and ataxia have been reported in approximately 55% of the dogs included in two studies evaluating the efficacy of GBP as adjunctive anti-epileptic treatment to PB and/or KBr (Govendir et al., 2005; Platt et al., 2006). As sedation and ataxia can be caused also by PB and KBr, concurrent administration of GBP may have been exacerbating rather than entirely causing these adverse effects. Sedation and ataxia were severe enough to warrant discontinuation of GBP in only one dog after the end of the study (Platt et al., 2006).
Although no adverse reactions have been reported in pharmacology studies and case reports in cats (Reid et al., 2010; Siao et al., 2010; Vettorato and Corletto, 2011; Lorenz et al., 2013; Steagall and Monteiro-Steagall, 2013), further data are needed on safety of chronic GBP administration in this species.
Dosing and Monitoring Recommendations
Based on the results of currently available pharmacologic studies (Radulovic et al., 1995; Siao et al., 2010; KuKanich and Cohen, 2011) and the target serum concentration in humans (>2 mg/ml) the recommended oral dosage of GBP is:
• 10–20 mg/kg body weight every 6 to 8 h
in dogs;
• 3–10 mg/kg every 6 to 8 h in cats.
Treatment can be started at the lower dose and eventually gradually increased based on clinical response and adverse effects.
As GBP clearance is highly correlated with renal function, dosage reduction is necessary in patients with reduced renal function (Bockbrader et al., 2010).
The range of serum or plasma GBP concentrations associated with seizure control in dogs and cats is unknown. In people, effective control of seizures typically requires serum or plasma GBP concentrations above 2 mg/ml (12 mmol/l) (Sivenius et al., 1991). An approximate reference range of 2–20 mg/ml (12–117 mmol/l) has been proposed (Johannessen et al., 2003; Lindberger et al., 2003; Patsalos et al., 2008), although a wide range of serum/plasma concentrations have been associated with clinical effect (Armijo et al., 2004; Patsalos et al., 2008) and serum concentrations at any given dose vary markedly between individuals (Gidal et al., 2000).
The main value of serum GBP concentration monitoring is to individualize the dose and dosing interval. As with other AEMs with a short half-life, sampling time in relation to dose administration is important for the interpretation of the drug concentration. Collection of a trough sample is recommended to evaluate the lowest concentration and to standardize sampling time in relation to dose.
In humans, if GBP discontinuation is desired, it has been recommended to taper the dose gradually over at least 1 week to avoid withdrawal symptoms, which may occur after as little as 1 month of treatment. Withdrawal symptoms may occur 1–3 days after abrupt discontinuation of GBP, particularly in patients administered high doses, and include mental status changes, anxiety, diaphoresis, palpitations, restlessness and rarely status epilepticus (Norton, 2001; Barrueto et al., 2002; Finch et al., 2010; See et al., 2011). Withdrawal symptoms resolve with re-initiation of GBP therapy.
Efficacy
In humans, GBP is considered efficacious as adjunctive and as monotherapy for focal seizures, however, PGB as well as other AEMs seem more efficacious than GBP (Silvenius et al., 1991; Marson et al., 2000, 2007; Yamauchi et al., 2006; Delahoy et al., 2010). GBP is not licensed for generalized seizure treatment and therefore information on efficacy for this seizure type is limited in humans. In one study, GBP resulted in greater reduction in the frequency of generalized tonic-clonic seizures than did placebo; however, the differences between treatments were not statistically significant. In addition, GBP did not affect the frequency of absence or myoclonic seizures (Chadwick et al., 1996).
The efficacy of GBP as adjunctive treatment to PB and/or KBr for canine idiopathic epilepsy has been investigated in two open uncontrolled clinical studies only to date (Govendir et al., 2005; Platt et al., 2006). The prospective study by Govendir et al. included 17 epileptic dogs (considered refractory to PB and/or KBr), which were administered GBP at 35 to 50 mg/kg/day (divided twice or three times daily) for at least 4 months. There was no significant decrease in overall mean monthly seizure frequency between baseline (before GBP initiation) and any of the 4 months of the study period. In addition, although three dogs had no seizures during the 4 months following GBP initiation, there was no statistically significant difference between the number of seizure-free dogs before and after initiation of GBP. Eleven (65%) dogs experienced a significant increase in the duration of the interictal period after GBP initiation. The limited efficacy of GBP in this study may have been due to the GBP administration interval of 12 h in the majority of dogs possibly resulting in sub-therapeutic serum GBP concentrations during part of the day (Govendir et al., 2005).
The study by Platt et al. included 11 idiopathic epileptic dogs with generalized onset seizures refractory to PB and KBr. Administration of GBP at 10 mg/kg every 8 h resulted in a significant decrease in the number of seizures per week and the number of days per week with any seizures when comparing the 3 months before and after initiation of GBP treatment. Six of 11 (55%) dogs had a reduction in the average number of seizures per week equal or greater than 50% (Platt et al., 2006).
To the best of the authors’ knowledge there are no studies on GBP efficacy as monotherapy in epileptic dogs and as mono- or adjunctive therapy in epileptic cats. In addition, the potential clinical benefits of the extended release GBP formulations currently approved for humans have not been evaluated in epileptic dogs and cats.
Pregabalin
PGB, (S)-3-aminomethyl-5-methylhexanoic acid, is a GABA analogue that is structurally
similar to GBP (Fig. 17.1). PGB was approved in 2004 by the European Agency for the Evaluation of Medicinal Products for treatment of adults with peripheral neuropathic pain and as adjunctive treatment for adults with focal seizures with or without secondary generalization. Subsequent approvals were granted in Europe for central neuropathic pain and for generalized anxiety disorders. The US FDA approved PGB in 2005 for neuropathic pain and adjunctive treatment for adults with focal seizures and in 2007 for management of fibromyalgia (Bockbrader et al., 2010). There are limited data on the use of PGB in dogs (Dewey et al., 2009; Salazar et al., 2009) and no information in cats.
Mechanism of Action
PGB mechanism of action remains incompletely elucidated. However, like GBP, PGB predominant pharmacological mechanism of action is mediated by binding to the alpha2-delta subunits of P/Q type voltage-gated calcium channels thereby selectively attenuating presynaptic calcium influx through these channels and decreasing the release of excitatory neurotransmitters such as glutamate, norepinephrine, serotonin, dopamine and substance P (Fig. 17.2) (Taylor et al., 2007; Schulze-Bonhage, 2013). PGB appears to have greater binding affinity for the alpha2-delta subunits than GBP (Li 2011). PGB does not interact with GABA receptors (Li et al., 2011).
Metabolism and Pharmacokinetics
Information on PBG pharmacokinetics in dogs is limited to one study whose results are illustrated in Table 17.3 (Salazar et al., 2009).
Additional studies are needed to investigate PGB metabolism and multiple administration pharmacokinetics in dogs. There are no data on PGB metabolism and pharmacokinetics in cats.
In humans PGB is rapidly absorbed (Tmax, 0.6–3.2 h) after oral ingestion with a bioavailability ³90%, which is independent of dose. CSF PGB concentration peaks 8 h following oral intake (Schulze-Bonhage, 2013). Co-administration of food decreases the rate but not the extent of PGB absorption (Bockbrader et al., 2010). Within the clinically used dose range, serum PGB concentrations are linearly related to dosage. Apparent volume of distribution is 0.5 l/kg, time to maximal plasma concentration (Cmax) is approximately 1 h, mean elimination half-life (T1/2) is 4.6–6.8 h and steady state is achieved within 24–48 h. PGB does not bind to plasma proteins and is excreted virtually unchanged (<2% metabolism) by the kidneys with a clearance that approximates glomerular filtration rate (Ben- Menachem, 2004; Bockbrader et al., 2010). PGB does not undergo hepatic metabolism and does not induce or inhibit
Table 17.3. Median and range pharmacokinetic parameters of PGB in healthy dogs administered a single oral dose of PGB (2 mg/kg or 4 mg/kg) (Salaza et al., 2009).
Pharmacokinetic parameters
Dose AUC Cmax T >2.8 administered(h·µg/ml)(µg/ml) Tmax (h) T1/2abs (h) T1/2 (h) µg/ml (h)
2 mg/kg PO Median 43.2 5 0.75 0.19 6.1 5.5 Minimum 41.2 4.1 0.5 0.17 5.79 5.2 Maximum 45.2 5.9 1 0.21 6.38 5.75
4 mg/kg PO Median 81.8 7.15 1.5 0.38 6.90 11.11 Minimum 56.5 4.6 1.0 0.25 6.21 6.97 Maximum 92.1 7.9 4.0 1.11 7.40 14.47
PO, per os; T1/2abs, absorption half-life; T1/2, elimination half-life; T >2.8 µg/ml, time over presumed minimal effective concentration; AUC, area under the curve from 0 to 36 h; C, maximal plasma concentration; T, time for C to occur
max maxmax
hepatic enzymes such as the cytochrome P450 system (Ben-Menachem, 2004).
Pharmacokinetic Interactions and Adverse Reactions
There is no information on PGB pharmacokinetic interactions in dogs and cats. As for GBP, the pharmacokinetic interaction potential of PGB is very low as PBG is not protein bound and does not induce nor inhibit hepatic CPY enzymes (Ben-Menachem, 2004; Pfizer, data on file, 2005). No clinically relevant pharmacokinetic drug interactions have been identified in humans to date (Perucca, 2006; Johannessen Landmark and Patsalos, 2010) with the exception of a moderate decrease (20% to 30%) in PGB concentrations following co-administration with enzyme-inducing AEMs such as carbamazepine, oxcarbazepine and phenytoin (May et al., 2007). PGB coadministration did not alter the steady-state plasma concentrations of PB, topiramate, carbamazepine, lamotrigine, phenytoin, tiagabine and valproate (Bockbrader et al., 2011). PGB has been shown to have some pharmacodynamic interactions such as additive effects on the cognitive and motor function impairment caused by oxycodone and potentiation of the CNS effects of lorazepam and ethanol (Ben-Menachem, 2004).
The most commonly reported adverse effects of PGB in humans are dose-related and include dizziness, somnolence and ataxia. These adverse effects are the most frequent reason for treatment discontinuation (Bockbrader et al., 2010; Schulze-Bonhage, 2013). Flexible dose titration minimizes these adverse effects following treatment initiation or dose increase. Other adverse effects include headache, dry mouth, peripheral oedema, blurred vision and dose-related weight gain (Stephen et al., 2011; Toth, 2012). When it occurs, weight gain is gradual over time and may be greater than 10% following 21 weeks of PGB treatment (Ryvlin et al., 2010). Somnolence has been reported to increase in humans co-prescribed antispasticity agents (Johannessen Landmark and Patsalos, 2010).
No adverse effects of PGB were observed in a pharmacokinetic study including healthy dogs administered a single oral dose of 4 mg/kg. In contrast, in a clinical study including 11 epileptic dogs administered PGB at 3 to 4 mg/kg orally every 8 h in combination with PB +/− KBr, sedation and ataxia were reported in ten (91%) dogs and persisted for the entire 3 month study period. In addition, one dog had episodes of apparent dizziness and weakness (Dewey et al., 2009).
Dosing and Monitoring Recommendations
Based on pharmacokinetic data from a study in clinically normal dogs and a presumed target range in humans of 2.8–8.2 mg/ml (Salazar et al., 2009), the proposed oral dose of PGB in dogs is:
• 3–4 mg/kg every 8–12 h.
To minimize PGB-related sedation and ataxia, it has been recommended to start PGB at 2 mg/kg orally every 8 to 12 h and increase it by 1 mg/kg each week until the target dose of 3–4 mg/kg is reached (Dewey et al., 2009). There are anecdotal reports of the use of PGB in cats, with a dose of 1–2 mg/kg orally every 12 h most commonly mentioned (Muñana, 2013). Further pharmacologic and clinical studies are required to identify the optimal dose and dosing interval for PGB.
As PGB clearance is highly correlated with renal function, dosage reduction is necessary in patients with impaired renal function (Ben-Menachem, 2004; Bockbrader et al., 2010). A 50% reduction in PGB daily dose has been recommended in people with creatinine clearance between 30 and 60 ml/ min compared to those with >60 ml/min (Randinitis et al., 2003).
No clear reference range has been established in epileptic humans, although an approximate range of 2.8–8.3 mg/ml has been reported (Berry and Millinigton, 2005; Patsalos et al., 2008).
A reference range for serum PGB concentrations associated with seizure control has not yet been established in dogs and cats. In the only clinical study in epileptic dogs administered PGB at 3–4 mg/kg orally every 8 h, trough serum PGB concentrations for all dogs ranged from 2.0 to 11.0 mg/ml (mean,
6.4 mg/ml; median, 7.3 mg/ml), however it was not possible to draw conclusions on association between seizure control and serum PGB concentrations (Dewey et al., 2009). As for GBP, if serum PGB monitoring is performed, it is preferable to obtain a trough sample to evaluate the lowest concentration and to standardize sampling time in relation to dose.
Efficacy
PGB anticonvulsant, analgesic and anxiolytic activities have been demonstrated in a range of animal models as well as in clinical studies in humans with focal seizures (French et al., 2003; Miller et al., 2003, Arroyo et al., 2004; Lee et al., 2009; Lozsadi et al., 2008), neuropathic pain (Dworkin et al., 2003; Pulman et al., 2014; Sabatowski et al., 2004; Freynhagen et al., 2005; Richter et al., 2005) and generalized anxiety disorder (Feltner et al., 2003; Pande et al., 2003; Rickels et al., 2005). Although there are no head-to-head randomized clinical trials comparing PGB and GBP, observational study data suggest that PGB is more potent and effective than GBP in the treatment of both refractory focal seizures and neuropathic pain (Bockbrader et al., 2010; Delahoy et al., 2010; Stephen et al., 2011). However, PGB efficacy as monotherapy in the treatment of focal seizures seems inferior to the efficacy of lamotrigine (Kwan et al., 2011).
The efficacy of PGB as adjunctive treatment to PB and/or KBr in idiopathic epileptic dogs has been investigated in one open-label non-comparative study, which included 11 dogs administered PGB at 3 to 4 mg/kg orally every 8 h (Dewey et al., 2009). The number of generalized seizures in the 3 months before and after initiation of PGB treatment was recorded and compared. Two dogs were prematurely withdrawn from the study at the request of the owners due to a combination of perceived lack of efficacy (139% increase in seizure frequency in one dog) and adverse effects. In the remaining nine dogs a significant reduction (mean, 57%; median, 50%) in seizure frequency was observed. Seven (64%) of the nine dogs that completed the study had a mean and median reduction in seizure frequency of 64% and 58%, respectively. Additionally, in seven dogs with cluster seizures, mean number of seizures for each cluster event was significantly decreased in the 3-month period after initiation of PGB treatment (Dewey et al., 2009).
No information is currently available on the efficacy of PGB in cats.
Summary Recommendations
• GBP and PGB have anti-epileptic and
antinociceptive effect in people and animal experimental models.
• PGB is considered more potent and
effective than GBP.
• GBP appears relatively well tolerated and
safe in dogs.
• PGB (3 to 4 mg/kg orally, every 8 h)
resulted in persistent sedation and/or ataxia in the majority of dogs included in a small clinical study and further investigation is required to evaluate PGB optimal dosage, tolerability and safety in epileptic dogs.
• GBP has a rapid elimination half-life and
therefore administration every 6 to 8 h is required; extended release formulations may overcome this problem.
• A reference range for serum GBP and PGB
concentrations associated with seizure control has yet to be identified in dogs and cats.
• Adjunctive treatment with GBP or PGB
may benefit some epileptic dogs, however further studies are needed to ascertain GBP and PGB efficacy as anti-epileptic medications in dogs.
• There is limited information on the use of
GBP in cats and further data are needed to evaluate its safety and efficacy.
• There are no data on tolerability, safety
and efficacy of PGB in cats.
References
Aghighi, S.A., Tipold, A., Piechotta, M., Lewczuk, P. and Kästner S.B. (2012) Assessment of the effects of adjunctive gabapentin on postoperative pain after intervertebral disc surgery in dogs. Veterinary Anaesthesia and Analgesia 39(6), 636–646.
Armijo, J.A., Perna, M.A., Adin, J. and Vega-Gil, N. (2004) Association between patient age and gabapentin serum concentration-to-dose ratio: a preliminary multivariate analysis. Therapeutic Drug Monitoring 26, 633–637.
Arroyo, S., Anhut, H., Kugler, A.R., Lee, C.M., Knapp, L.E., Garofalo, E.A. and Messmer, S. (2004) Pregabalin 1008-011 International Study Group. Pregabalin add-on treatment: a randomized, double-blind, placebo-controlled, dose-response study in adults with partial seizures. Epilepsia 45(1), 20–27.
Backonja, M. and Glanzman, R.L. (2003) Gabapentin dosing for neuropathic pain: evidence from randomized, placebo-controlled clinical trials. Clinical Therapeutics 25, 81–104.
Barrueto, F. Jr, Green, J., Howland, M.A., Hoffman, R.S. and Nelson, L.S. (2002) Gabapentin withdrawal presenting as status epilepticus. Journal of Toxicology, Clinical Toxicolology 40(7), 925–928.
Ben-Menachem, E. (2004) Pregabalin pharmacology and its relevance to clinical practice. Epilepsia 45(Suppl. 6), 13–18.
Bennett, M.I. and Simpson, K.H. (2004) Gabapentin in the treatment of neuropathic pain. Palliative Medicine 18(1), 5–11.
Berry, D. and Millinigton, C. (2005) Analysis of pregabalin at therapeutic concentrations in human plasma/ serum by reversed-phased HPLC. Therapeutic Drug Monitoring 27, 451–456.
Berry, D.J., Beran, R.G., Plunkeft, M.J., Clarke, L.A. and Hung, W.T. (2003) The absorption of gabapentin following high dose escalation. Seizure 12, 28–36.
Bertrand, S., Ng, G.Y.K., Purisai, M.G., Wolfe, S.E., Severidt, M.W., Nouel, D., Robitaille, R., Low, M.J., O’Neill, G.P., Metters, K., Lacaille, J.C., Chronwall, B.M. and Morris, S.J. (2001) The anticonvulsant, antihyperalgesic agent gabapentin is an agonist at brain g-aminobutyric acid type B receptors negatively coupled to voltage-dependent calcium channels. The Journal of Pharmacology and Experimental Therapeutics 298, 15–24.
Bertrand, S., Nouel, D., Morin, F., Nagy, F. and Lacaille, J.C. (2003) Gabapentin actions on Kir3 currents and N-type Ca2+ channels via GABAB receptors in hippocampal pyramidal cells. Synapse 50, 95–109.
Bockbrader, H.N., Wesche, D., Miller, R., Chapel, S., Janiczek, N. and Burger, P. (2010) A comparison of the pharmacokinetics and pharmacodynamics of pregabalin and gabapentin. Clinical Pharmacokinetics 49, 661–669.
Bockbrader, H.N., Burger, P. and Knapp, L. (2011) Pregabalin effect on steady-state pharmacokinetics of carbamazepine, lamotrigine, phenobarbital, phenytoin, topiramate, valproate, and tiagabine. Epilepsia 52, 405–409.
Cashmore, R.G., Harcourt-Brown, T.R., Freeman, P.M., Jeffery, N.D. and Granger, N. (2009) Clinical diagnosis and treatment of suspected neuropathic pain in three dogs. Australian Veterinarian Journal 87, 45–50.
Chadwick, D.W., Leiderman, D.B., Sauermann, W., Alexander, J. and Garofalo, E. (1996) Gabapentin in generalized seizures. Epilepsy Research 25, 191–197.
Cheng, J.K. and Chiou, L.C. (2006) Mechanisms of the antinociceptive action of gabapentin. Journal of Pharmacological Science 100(5), 471–486. Epub Feb 11.
Clarke, H., Bonin, R.P., Orser, B.A., Englesakis, M., Wijeysundera, D.N. and Katz, J. (2012) The prevention of chronic postsurgical pain using gabapentin and pregabalin: a combined systematic review and metaanalysis. Anesthesia and Analgesia 115(2), 428–442. Epub 2012 Mar 13.
Coderre, T.J., Kumar, N., Lefebvre, C.D. and Yu, J.S. (2005) Evidence that gabapentin reduces neuropathic pain by inhibiting the spinal release of glutamate. Journal of Neurochemistry 94, 1131–1139.
Cundy, K.C., Sastry, S., Luo, W., Zou, J., Moors, T.L. and Canafax, D.M. (2008) Clinical pharmacokinetics of XP13512, a novel transported prodrug of gabapentin. Journal of Clinical Pharmacology 48(12), 1378–1388.
Cunningham, M.O., Woodhall, G.L., Thompson, S.E., Dooley, D.J. and Jones R.S.G. (2004) Dual effects of gabapentin and pregabalin on glutamate release at rat entorhinal synapses in vitro. European Journal of Neuroscience 20, 1566–1576.
Delahoy, P., Thompson, S. and Marschner, I.C. (2010) Pregabalin versus gabapentin in partial epilepsy: a meta-analysis of dose-response relationships. BMC Neurology 10, 104.
Dewey, C.W., Cerda-Gonzalez, S. and Levine, J.M. (2009) Pregabalin as an adjunct to phenobarbital, potassium bromide, or a combination of phenobarbital and potassium bromide for treatment of dogs with suspected idiopathic epilepsy. Journal of American Veterinary Medical Association 235, 1442–1449.
Dworkin, R.H., Corbin, A.E., Young, J.P., Sharma, U., LaMoreaux, L., Bockbrader, H., Garofalo, E.A. and Poole, R.M. (2003) Pregabalin for the treatment of postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology 60, 1274–1283.
Eroglu, C., Allen, N.J. and Susman, M.W. (2009) Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392.
Feltner, D.E., Crockatt, J.G., Dubovsky, S.J., Cohn, C.K., Shrivastava, R.K., Targum, S.D., Liu-Dumaw, M., Carter, C.M. and Pande, A.C. (2003) A randomized, double-blind, placebo-controlled, fixed-dose, multicenter study of pregabalin in patients with generalized anxiety disorder. Journal of Clinical Psychopharmacology 23, 240–249.
Field, M.J., Cox, P.J., Stott, E., Melrose, H., Offord, J., Su, T.Z., Bramwell, S., Corradini, L., England, S., Winks, J., Kinloch, R.A., Hendrich, J., Dolphin, A.C., Webb, T. and Williams, D. (2006) Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proceedings of the National Academy of Sciences of the United States of America 103, 17537–17542.
Finch, C.K., Eason, J. and Usery, J.B. (2010) Gabapentin withdrawal syndrome in a post-liver transplant patient. Journal of Pain and Palliative Care Pharmacotherapy 24(3), 236–238.
French, J.A., Kugler, A.R., Robbins, J.L., Knapp, L.E. and Garofalo, E.A. (2003) Dose response trial of pregabalin adjunctive therapy in patients with partial seizures. Neurology 60, 1631–1637.
Freynhagen, R., Strojek, K., Griesing, T., Whalen, E. and Balkenohl, M. (2005) Efficacy of pregabalin in neuropathic pain evaluated in a 12-week, randomised, double-blind, multicentre, placebo- controlled trial of flexible- and fixed-dose regimens. Pain 115, 254–263.
Gidal, B.E., Radulovic, L.L., Kruger, S., Rutecki, P., Pitterle, M. and Bockbrader, H.N. (2000) Inter- and intra-subject variability in gabapentin absorption and absolute bioavailability. Epilepsy Research 40, 123–127.
Gordi, T., Hou, E., Kasichayanula, S. and Berner, B. (2008) Pharmacokinetics of gabapentin after a single day and at steady state following the administration of gastric-retentive extended-release and immediate-release tablets: a randomized, open-label, multiple-dose, three-way crossover, exploratory study in healthy subjects. Clinical Therapeutics 30, 909–916.
Govendir, M., Perkins, M. and Malik, R. (2005) Improving seizure control in dogs with refractory epilepsy using gabapentin as an adjunctive agent. Australian Veterinary Journal 83, 602–608.
Hooper, W.D., Kavanagh, M.C., Herkes, G.K. and Eadie, M.J. (1991) Lack of a pharmacokinetic interaction between phenobarbitone and gabapentin. British Journal of Clinical Pharmacology 31(2), 171–174.
Hussein, G., Troupin, A.S. and Montouris, G. (1996) Gabapentin interaction with felbamate. Neurology 47(4), 1106.
Jensen, A.A., Mosbacher, J., Elg, S., Lingenhoehl, K., Lohmann, T., Johansen, T.N., Abrahamsen, B., Mattsson, J.P., Lehmann, A., Bettler, B. and Bräuner-Osborne, H. (2002) The anticonvulsant gabapentin (neurontin) does not act through gamma-aminobutyric acid-B receptors. Molecular Pharmacology 61, 1377–1384.
Johannessen, S.I., Battino, D., Berry, D.J., Bialer, M., Kramer, G., Tomson, T. and Patsalos, P.N. (2003) Therapeutic drug monitoring of the newer antiepileptic drugs. Therapeutic Drug Monitoring 25, 347–363.
Johannessen Landmark, C. and Patsalos, P.N. (2010) Drug interactions involving the new second- and third-generation antiepileptic drugs. Expert Review of Neurotherapeutics 10(1), 119–140.
Kim, Y.S., Chang, H.K., Lee, J.W., Sung, Y.H., Kim, S.E., Shin, M.S., Park, J.H., Kim, H. and Kim, C.J. (2009) Protective effect of gabapentin on N-methyl- D-aspartate-induced excitotoxicity in rat hippocampal CA1 neurons. Journal of Pharmacological Sciences 109, 144–147.
KuKanich, B. and Cohen, R.L. (2011) Pharmacokinetics of oral gabapentin in greyhound dogs. Veterinary Journal 187, 133–135.
Kwan, P., Brodie, M.J., Kälviäinen, R., Yurkewicz, L., Weaver, J. and Knapp, L.E. (2011) Efficacy and safety of pregabalin versus lamotrigine in patients with newly diagnosed partial seizures: a phase 3, double-blind, randomised, parallel-group trial. Lancet Neurology 10(10), 881–890.
Lamont, L. (2008) Adjunctive analgesic therapy in veterinary medicine. Veterinary Clinics of North America Small Animal Practice 38, 1187–1203.
LaRoche, S.M. and Helmers, S.L. (2004) The new antiepileptic drugs: scientific review. JAMA: The Journal of the American Medical Association 291(5), 605–614.
Lee, B.I., Yi, S., Hong, S.B., Kim, M.K., Lee, S.A., Lee, S.K., Shin, D.J., Kim, J.M., Song, H.K., Heo, K., Lowe, W. and Leon, T. (2009) Pregabalin add-on therapy using a flexible, optimized dose schedule in refractory partial epilepsies: a double-blind, randomized, placebo-controlled, multicenter trial. Epilepsia 50(3), 464–474.
Li, Z., Taylor, C.P., Weber, M., Piechan, J., Prior, F., Bian, F., Cui, M., Hoffman, D. and Donevan, S. (2011) Pregabalin is a potent and selective ligand for a(2)d-1 and a(2)d-2 calcium channel subunits. European Journal of Pharmacology 667(1–3), 80–90.
Lindberger, M., Luhr, O., Johannessen, S.I., Larsson, S. and Tomson, T. (2003) Serum concentrations and effects of gabapentin and vigabatrin: observations from a dose titration study. Therapeutic Drug Monitoring 25, 457–462.
Lorenz, N.D., Comerford, E.J. and Iff, I. (2012) Long-term use of gabapentin for musculoskeletal disease and trauma in three cats. Journal of Feline Medicine and Surgery 15(6), 507–512.
Lozsadi, D., Hemming, K. and Marson, A.G. (2008) Pregabalin add-on for drug-resistant partial epilepsy. Cochrane Database System Reviews Jan 23; (1): CD005612.
Maneuf, Y.P., Hughes, J. and McKnight, A.T. (2001) Gabapentin inhibits the substance P-facilitated K+-evoked release of [3H]glutamate from rat caudal trigeminal nucleus slices. Pain 93, 191–196.
Maneuf, Y.P., Blake, R., Andrews, N.A. and McKnight, A.T. (2004) Reduction by gabapentin of K+-evoked release of [3H]-glutamate from the caudal trigeminal nucleus of the streptozotocin-treated rat. British Journal of Pharmacology 141, 574–579.
Marson, A.G., Kadir, Z.A., Hutton, J.L. and Chadwick, D.W. (2000) Gabapentin add-on for drug-resistant partial epilepsy. Cochrane Database of Systematic Reviews (3): CD001415.
Marson, A.G., Al-Kharusi, A.M., Alwaidh, M., Appleton, R., Baker, G.A., Chadwick, D.W., Cramp, C., Cockerell, O.C., Cooper, P.N., Doughty, J., Eaton, B., Gamble, C., Goulding, P.J., Howell, S.J., Hughes, A., Jackson, M., Jacoby, A., Kellett, M., Lawson, G.R., Leach, J.P., Nicolaides, P., Roberts, R., Shackley, P., Shen, J., Smith, D.F., Smith, P.E., Smith, C.T., Vanoli, A., Williamson, P.R. and SANAD Study Group (2007) The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 369, 1000–1015.
Matar, K.M., Nicholis, P., Bawazir, S.A., Al-Hassan, M.I., Tekle, A., Bawazir, S.A. and al-Hassan, M.I. (2000) Effect of vigabatrin and gabapentin on phenytoin pharmacokinetics in the dog. European Journal of Drug Metabolism and Pharmacokinetics 42, 517–521.
May, T.W., Rambeck, B., Neb, R. and Jurgens, U. (2007) Serum concentrations of pregabalin in patients with epilepsy: the influence of dose, age, and comedication. Therapeutic Drug Monitoring 29, 789–794.
McClelland, D., Evans, R.M., Barkworth, L., Martin, D.J. and Scott, R.H. (2004) A study comparing the actions of gabapentin and pregabalin on the electrophysiological properties of cultured DRG neurons from neonatal rats. BMC Pharmacology 4, 14.
McLean, M.J. (1995) Gabapentin. Epilepsia 36(Suppl. 2), S73–S86.
Miller, R., Frame, B., Corrigan, B., Burger, P., Bockbrader, H., Garofalo, E. and Lalonde, R. (2003) Exposure-response analysis of pregabalin add-on treatment of patients with refractory partial seizures. Clinical Pharmacology and Therapeutics 73, 491–505.
Muñana, K.R. (2013) Update: seizure management in small animal practice. Veterinary Clinics of North America Small Animal Practice 43(5), 1127–1147.
Norton, J.W. (2001) Gabapentin withdrawal syndrome. Clinical Neuropharmacology 24(4), 245–246.
Pande, A.C., Crockatt, J.G., Feltner, D.E., Janney, C.A., Smith, W.T., Weisler, R., Londborg, P.D., Bielski, R.J., Zimbroff, D.L., Davidson, J.R.T. and Liu-Dumaw, M. (2003) Pregabalin in generalized anxiety disorder: a placebo-controlled trial. The American Journal of Psychiatry 160, 533–540.
Pandey, C.K., Singhal, V., Kumar, M., Lakra, A., Ranjan, R., Pal, R., Raza, M., Singh, U. and Singh, P.K. (2005) Gabapentin provides effective postoperative analgesia whether administered pre-emptively or post-incision. Canadian Journal of Anaesthesia 52, 827–831.
Parker, D.A.S., Ong, J., Marino, V. and Kerr, D.I.B. (2004) Gabapentin activates presynaptic GABAB heteroreceptors in rat cortical slices. European Journal of Pharmacology 495, 137–143.
Patsalos, P.N., Berry, D.J., Bourgeois, B.F., Cloyd, J.C., Glauser, T.A., Johannessen, S.I., Leppik, I.E., Tomson, T. and Perucca, E. (2008) Antiepileptic drugs–best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 49, 1239–1276.
Perucca, E. (2006) Clinically relevant drug interactions with antiepileptic drugs. British Journal of Clinical Pharmacology 61(3), 246–255.
Petroff, O.A.C., Rothman, D.L., Behar, K.L., Lamoureux, D. and Mattson, R.H. (1996) The effect of gabapentin on brain gamma-aminobutyric acid in patients with epilepsy. Annals of Neurology 39, 95–99.
Platt, S.R., Adams, V., Garosi, L.S., Abramson, C.J., Penderis, J., De Stefani, A. and Matiasek, L. (2006) Treatment with gabapentin of 11 dogs with refractory idiopathic epilepsy. Veterinary Record 159, 881–884.
Pulman, J., Hemming, K., Marson, A.G. (2014) Pregabalin add-on for drug-resistant partial epilepsy. The Cochrane Database of Systematic Reviews: CD005612.
Radulovic, L.L., Türck, D., Von Hodenberg, A., Vollmer, K.O., McNally, W.P., DeHart, P.D., Hanson, B.J., Bockbrader, H.N. and Chang, T. (1995) Disposition of gabapentin (neurontin) in mice, rats, dogs, and monkeys. Drug Metabolism and Disposition 23, 441–448.
Randinitis, E.J., Posvar, E.L., Alvey, C.W., Sedman, A.J., Cook, J.A. and Bockbrader, H.N. (2003) Pharmacokinetics of pregabalin in subjects with various degrees of renal function. Journal of Clinical Pharmacology 43, 277–283.
Reid, P., Pypendop, B.H. and Ilkiw, J.E. (2010) The effects of intravenous gabapentin administration on the minimum alveolar concentration of isoflurane in cats. Anesthesia and Analgesia 111(3), 633–637.
Rhee, Y.S., Park, S., Lee, T.W., Park, C.W., Nam, T.Y., Oh, T.O., Jeon, J.W., Han, S.B., Lee, D.S. and Park, E.S. (2008) In Vitro/in vivo relationship of gabapentin from a sustained-release tablet formulation: a pharmacokinetic study in the beagle dog. Archives of Pharmacology Research 31, 911–917.
Richter, R.W., Portenoy, R., Sharma, U., Lamoreaux, L., Bockbrader, H. and Knapp, L.E. (2005) Relief of painful diabetic peripheral neuropathy with pregabalin: a randomized, placebo-controlled trial. The Journal of Pain: the Official Journal of the American Pain Society 6, 253–260.
Rickels, K., Pollack, M.H., Feltner, D.E., Lydiard, R.B., Zimbroff, D.L., Bielski, R.J., Tobias, K., Brock, J.D., Zornberg, G.L. and Pande, A.C. (2005) Pregabalin for treatment of generalized anxiety disorder: a 4-week, multicenter, double-blind, placebo-controlled trial of pregabalin and alprazolam. Archives of General Psychiatry 62, 1022–1030.
Rosner, H., Rubin, L. and Kestenbaum, A. (1996) Gabapentin adjunctive therapy in neuropathic pain states. The Clinical Journal of Pain 12, 56–58.
Ryvlin, P., Kälviäinen, R., Von Raison, F., Giordano, S., Emir, B. and Chatamra, K. (2010) Pregabalin in partial seizures: a pragmatic 21-week, open-label study (PREPS). European Journal of Neurology 17(5), 726–732.
Sabatowski, R., Galvez, R., Cherry, D.A., Jacquot, F., Vincent, E., Maisonobe, P., Versavel, M. and the 1008-045 Study Group (2004) Pregabalin reduces pain and improves sleep and mood disturbances in patients with post-herpetic neuralgia: results of a randomised, placebo-controlled clinical trial. Pain 109, 26–35.
Salazar, V., Dewey, C.W., Schwark, W., Gleed, R.D., Horne, W. and Ludder, J.W. (2009) Pharmacokinetics of single-dose oral pregabalin administration in normal dogs. Veterinary Anaesthesia and Analgesia 36, 574–580.
Schulze-Bonhage, A. (2013) Pharmacokinetic and pharmacodynamic profile of pregabalin and its role in the treatment of epilepsy. Expert Opinion Drug Metabolism and Toxicology 9(1), 105–115.
See, S., Hendriks, E. and Hsiung, L. (2011) Akathisia induced by gabapentin withdrawal. The Annals of Pharmacotherapy 45(6): e31. Epub 2011 Jun 7.
Siao, K.T., Pypendop, B.H. and Ilkiw, J.E. (2010) Pharmacokinetics of gabapentin in cats. American Journal of Veterinary Research 71, 817–821.
Sills, G. (2006) The mechanisms of action of gabapentin and pregabalin. Current Opinion in Pharmacology 6, 108–113.
Sivenius, J., Kälviäinen, R., Ylinen, A. and Riekkinen, P. (1991) Double-blind study of gabapentin in the treatment of partial seizures. Epilepsia 32, 539–542.
Steagall, P.V. and Monteiro-Steagall, B.P. (2013) Multimodal analgesia for perioperative pain in three cats. Journal of Feline Medicine and Surgery, Feb 4. [Epub ahead of print]
Stephen, L.J., Parker, P., Kelly, K., Wilson, E.A., Leach, V. and Brodie M.J. (2011) Adjunctive pregabalin for uncontrolled partial-onset seizures: findings from a prospective audit. Acta Neurologica Scandinavica 124(2), 142–145.
Stewart, B.H., Kugler, A.R., Thompson, P.R. and Bockbrader, H.N. (1993) A saturable transport mechanism in the intestinal absorption of gabapentin is the underlying cause of the lack of proportionality between increasing dose and drug concentrations in plasma. Pharmaceutical Research 10(2), 276–281.
Suarez, L.M., Suarez, F., Del Olmo, N., Ruiz, M., Gonzalez-Escalada, J.R. and Solis, J.M. (2005) Presynaptic NMDA autoreceptors facilitate axon excitability: a new molecular target for the anticonvulsant gabapentin. European Journal of Neuroscience 21, 197–209.
Taylor, C.P., Gee, N.S., Su, T.Z., Kocsis, J.D., Welty, D.F., Brown, J.P., Doodley, D.J., Boden, P. and Singh, L. (1998) A summary of mechanistic hypotheses of gabapentin pharmacology. Epilepsy Research 29, 233–249.
Taylor, C.P., Angelotti, T. and Fauman, E. (2007) Pharmacology and mechanism of action of pregabalin: the calcium channel alpha2-delta (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy Research 73(2), 137–150.
Toth, C. (2012) Drug safety evaluation of pregabalin. Expert Opinion on Drug Safety 11(3), 487–502.
Vettorato, E. and Corletto, F. (2011) Gabapentin as part of multi-modal analgesia in two cats suffering multiple injuries. Veterinary Anaesthesia and Analgesia 38, 518–520.
Vollmer, K.O., Von Hodenberg, A. and Kölle, E.U. (1986) Pharmacokinetics and metabolism of gabapentin in rat, dog and man. Arzneimittelforschung 36, 830–839.
Wagner, A.E., Mich, P.M., Uhrig, S.R. and Hellyer, P.W. (2010) Clinical evaluation of perioperative administration of gabapentin as an adjunct for postoperative analgesia in dogs undergoing amputation of a forelimb. Journal of American Veterinary Medical Association 236, 751–756.
Wolfe, K. and Poma, R. (2010) Syringomyelia in the Cavalier King Charles spaniel (CKCS) dog. Canine Veterinary Journal 51, 95–102.
Yamauchi, T., Kaneko, S., Yagi, K. and Sase, S. (2006) Treatment of partial seizures with gabapentin: double-blind, placebo-controlled, parallel-group study. Psychiatry and Clinical Neuroscience 60(4), 507–515.
18 Felbamate
Simon Platt BVM&S MRCVS Dipl. ACVIM (Neurology) Dipl. ECVN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
Felbamate was the first of the new wave antiseizure medications introduced in the 1990s and the first since 1978. Based on the information available in the human literature, this medication has a broad spectrum of activity on multiple seizure types, particularly in refractory epileptics (Pellock, 1999). Limited information is available on its use clinically in veterinary medicine, although one study described later in this chapter has determined it to be successful at least for focal seizure activity in dogs (Ruehlmann et al., 2001). Felbamate is a dicarbamate (2-phenyl1,3-propanediol dicarbamate) and resembles the minor tranquilizer meprobamate, although it has minimal sedative or tranquilizing properties at therapeutic levels (Fig. 18.1; Bourgeois, 1997). The neurotoxicity of this medication in rodent models is low, and repeated administration in various species initially suggested an excellent safety profile (Pellock, 1999). The main indication in humans for the use of this drug is in children with Lennox-Gastaut syndrome (LGS) who have failed to respond to other anti-epileptic drugs such as clonazepam and valproate. LGS, also known as Lennox syndrome, is a difficult-to-treat form of childhood-onset epilepsy that most often appears between the second and sixth year of life, and is characterized by frequent seizures and different seizure types; it is often accompanied by developmental delay and psychological and behavioural problems. Based on the aforementioned information, felbamate is currently considered as a fourth or fifth drug in patients with refractory onset seizures, especially in children (Pellock, 1999).
Mechanism of Action
No single mechanism for the anti-epileptic effect of felbamate has been identified, but several actions have been shown. Felbamate was shown to inhibit glycine-enhanced NMDA-induced intracellular calcium currents in mice (White et al., 1995). Contradictory findings have been reported on the effect of felbamate on the gamma-aminobutyric acid (GABA) receptor; the relatively minor effect on this receptor may underlie the lack of sedative effects and absence of adverse effects on cognition associated with felbamate (Sankar and Holmes, 2004). Both a lack of effect on ligand binding to the receptor as well as potentiation of GABA responses at high felbamate concentrations have been reported in people (Ticku et al., 1991; Rho et al., 1994; Bourgeois, 1997). Additionally, in vitro work has shown that at high felbamate concentrations, inhibition of excitatory NMDA responses was shown, making it the first anti-epileptic drug with dual action on the excitatory and inhibitory
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
brain transmission (Rho et al., 1994). Other in vitro work has established that blockade of voltage-gated sodium channels and inhibition of voltage-gated calcium currents are mechanisms of action for this drug (Fig. 18.2; White, 1999; Pellock et al., 2006).
In rodent models, felbamate is effective at non-toxic doses in preventing NMDA agonist-induced seizures and stopping supramaximal tonic extensor seizures and induced by pentylenetetrazol or picrotoxin (Ruehlmann et al., 2001). This medication has also been shown to block corneal-kindled seizures in rats, a supposed accurate indicator of efficacy in the treatment of focal seizures (Loscher and Schmidt, 1988; Bourgeois, 1997).
 O NH2
O NH2
O
O NH2
O
Metabolism and Pharmacokinetics
At least 90% of an oral dose is absorbed based on adult human and dog studies with single dose and chronic administration studies suggesting that the absorption is linear (Bourgeois, 1997). Following an oral dose, time to peak concentration varies between 2 and 6 h in humans and dogs. Approximately 25% of the dose binds to proteins. In humans and dogs, approximately 50–76% of a felbamate dose is excreted in the urine unmetabolized and unconjugated (Yang et al., 1991; Bourgeois, 1997). Approximately 12% is excreted in the urine as para-hydroxyfelbamate and 2-hydroxyfelbamate. Most of the remainder of the dose is recovered in urine as unidentified polar metabolites, some of them as glucuronides or sulfate esters. The elimination half-life ranges from 16 to 24 h in humans and 4 to 6 h in dogs (Adusumalli et al., 1992). In dogs, this results in a steady-state concentration in 20–30 h. Felbamate is partially metabolized by the P450 system, especially CYP3A4, via hydroxylation and conjugation (Pellock et al., 2006).
Penetration of felbamate into the brain is excellent as it is lipophilic, with up to 70%

Fig. 18.2. The sites of action of felbamate at the neuronal synapse are highlighted.
Felbamate 455
of plasma concentrations being detected in humans (Adusumalli et al., 1994).
Felbamate plasma concentrations have been determined in male and female adult and immature beagle dogs after a single and ten once per day oral doses of 60 mg/kg of drug (Adusumalli et al., 1992; Yang et al., 1992). No gender-related differences were noted; however, the bioavailability of felbamate in immature dogs was threefold lower than in adult dogs (Adusumalli et al., 1992). More rapid overall urinary elimination of the drug in immature dogs due to an increased rate of metabolism appears to be responsible for the lower bioavailability (Yang et al., 1992). Currently, there are no available data on the metabolism and pharmacokinetics of felbamate in cats.
Pharmacokinetic Interactions and Adverse Reactions
Pharmacokinetic interactions between felbamate and other anti-epileptic medications have been well studied. Felbamate is both the cause and object of several pharmacokinetic interactions in people. Examples of this include raising concurrent phenobarbital, phenytoin and valproate serum levels in a dose-dependent manner, while reducing carbamazepine levels (Bourgeois, 1997). The elimination of felbamate was noted to be strikingly reduced when given with gabapentin (Hussein et al., 1996). This interaction represents an inhibitory effect on elimination itself and not a metabolic interaction, presumably due to active competition within the kidney.
The elimination of felbamate is affected by enzyme inducers, increasing the total body clearance when given concurrently, necessitating increased dosages to achieve the required serum levels (Wagner et al., 1991). In general, felbamate acts as an enzyme inhibitor rather than as an enzyme inducer; thus, inhibition of CYP2C19 results in a 30–50% rise in serum levels of drugs which are metabolized by this enzyme when felbamate is added (Pellock et al., 2006). Phenobarbital levels increase by a mean of 24% when felbamate is added and so a dose reduction of phenobarbital would be indicated if combination therapy were being considered (Reidenberg et al., 1995). Diazepam is affected similarly by this inhibition.
When this drug was initially released in 1993, it was marketed as a safe antiseizure medication, which lacked demonstrable toxic side effects and did not require any form of laboratory monitoring. Within a year of its release, it became evident that the known side-effects were greater than anticipated but that felbamate was associated with an unacceptable incidence of life-threatening side-effects (Bourgeois, 1997). Adverse side-effects were reported in 24% of children in one study (Pellock et al., 2006). Gastrointestinal side-effects including anorexia, 3–5% weight loss and vomiting were noted in one out of ten human patients and were more likely when the drug was used as an adjunctive therapy. Insomnia and irritability were also noted to be significant and headaches affected 36–40% of people on the drug (Bourgeois, 1997). The medication was nearly withdrawn in 1994 because of the incidence of aplastic anaemia and fatal hepatotoxicity. The risk of aplastic anaemia associated with this medication is estimated to be about 12 per 100,000 patients, with risk factors for development being listed as previous history of cytopenias and history of allergy (Kaufman et al., 1997; Pellock et al., 2006). The risk of associated death from one of these as yet idiosyncratic complications was calculated to be 1:5500 patients (White et al., 1995).
In a clinical trial evaluating felbamate for the control of focal seizures in six dogs, the side effects noted included keratitis sicca, which progressed to corneal perforation in one dog (Ruehlmann et al., 2001). Sedation and weakness was not seen in any of the dogs. Two of the six dogs experienced mild but non-lifethreatening blood dyscrasias, including thrombocytopenia (13 months after initiating the drug), and lymphopenia and leucopenia (3 months after drug initiation).
Dosing and Monitoring Recommendations
In humans, on account of the serious side-effects that were noted, a titrated dose
introduction is advised over a 3-week period with a reduction in concurrent antiseizure medications by approximately 25% (Pellock, 1999). Withdrawal, if necessary, should also be performed in a tapered fashion where possible as status epilepticus has occurred in humans following rapid cessation of felbamate therapy (DeGiorgio et al., 1995).
Doses in dogs ranging from 62 to 220 mg/kg/day (median 77) divided two to three times daily and administered orally have been reported (Adusumalli et al., 1992). Dosing three to four times daily has been recommended in adult dogs whilst it may be necessary to administer even more frequently in immature dogs. The recommended initial starting dose is 20 mg/kg orally three times daily, increasing to 400–600 mg/day every 1 to 2 weeks in adult dogs (Adusumalli et al., 1992). An upper limit dose based on published safety studies has not been established in the dog.
Haematologic evaluations should be performed before felbamate therapy is initiated, during therapy, and after discontinuation. Patients should be monitored frequently through clinical and laboratory means. In humans, the signs of aplasic anaemia and liver failure are usually seen during the first 6–12 months of therapy. A minimum of monthly blood tests should be performed for this period of time, following up every 6–12 months after this. Serum felbamate concentrations can be assessed routinely in dogs using HPLC (Clark et al., 1992; Romanyshyn et al., 1993).
Efficacy
Based on human trials, felbamate is effective in patients with focal epilepsy as well as absence seizures and juvenile myoclonic epilepsy (Pellock, 1999). As an adjunctive drug in people, felbamate was reported to at least halve the seizure frequency in 16–20% of adults and 53% of children (Carmant et al., 1994; Li et al., 1996). However, a recent Cochrane review looking at the efficacy of felbamate for refractory focal-onset epilepsy found only three randomized trials investigating this subject and could not conclude that there was any evidence to support the use of felbamate in these circumstances (Shi et al., 2011).
The only efficacy data in veterinary medicine evaluating felbamate investigated its use in six dogs with refractory focal seizure activity due to an idiopathic aetiology (Ruehlmann et al., 2001). Three dogs were administered felbamate as a monotherapy, and in three dogs felbamate was added to a prior phenobarbital regimen. The duration of therapy ranged from 2 to 22 months and the dose ranged from 62 to 220 mg/kg/day (median 77). All six dogs in the study experienced a reduction in their seizure frequency shortly after felbamate therapy was started or adjusted in its dose. Seizures were completely eliminated in two dogs, with a reduction in seizure frequency and severity in the remaining four dogs. No correlation was found between serum levels of the drug and efficacy although a therapeutic window for felbamate in humans has been calculated to be 50–110 mg/l (Ruehlmann et al., 2001). No data on the efficacy or even use of this drug in cats could be found at the time of writing.
Summary Recommendations
• Felbamate should be reserved for dogs
refractory to the other more thoroughly investigated and safer antiepileptic medications in this species and as such this is a fourth or fifth line option.
• Felbamate may be most appropriate in
dogs with focal seizure activity.
• Close attention needs to be paid both to
clinical signs and regular blood work for the potential of bone marrow and liver toxicity.
• This drug should not be used in dogs
with pre-existing hepatic disease.
• Serum therapeutic levels do not correlate with seizure control and so blood samples may not be necessary in most dogs.
• Caution should be used when administering felbamate in combination with phenobarbital, as potentially toxic concentrations of phenobarbital may result.
Felbamate 457
References
Adusumalli, V.E., Gilchrist, J.R., Wichmann, J.K., Kucharczyk, N. and Sofia, R.D. (1992) Pharmacokinetics of felbamate in pediatric and adult beagle dogs. Epilepsia 33, 955–960.
Adusumalli, V.E., Wichmann, J.K., Kucharczyk, N., Kamin, M., Sofia, R.D., French, J., Sperling, M., Bourgeois, B., Devinsky, O., Dreifuss, F.E. et al. (1994) Drug concentrations in human brain tissue samples from epileptic patients treated with felbamate. Drug Metabolism and Disposition 22, 168–170.
Bourgeois, B.F. (1997) Felbamate. Seminars in Pediatric Neurology 4, 3–8.
Carmant, L., Holmes, G.L., Sawyer, S., Rifai, N., Anderson, J. and Mikati, M.A. (1994) Efficacy of felbamate in therapy for partial epilepsy in children. Journal of Pediatrics 125, 481–486.
Clark, L.A., Wichmann, J.K., Kucharczyk, N. and Sofia, R.D. (1992) Determination of the anticonvulsant felbamate in beagle dog plasma by high-performance liquid chromatography. Journal of Chromatography 573, 113–119.
DeGiorgio, C.M., Lopez, J.E., Lekht, Z.N. and Rabinowicz, A.L. (1995) Status epilepticus induced by Felbatol withdrawal. Neurology 45, 1021–1022.
Hussein, G., Troupin, A.S. and Montouris, G. (1996) Gabapentin interaction with felbamate. Neurology 47, 1106.
Kaufman, D.W., Kelly, J.P., Anderson, T., Harmon, D.C. and Shapiro, S. (1997) Evaluation of case reports of aplastic anemia among patients treated with felbamate. Epilepsia 38, 1265–1269.
Li, L.M., Nashef, L., Moriarty, J., Duncan, J.S. and Sander, J.W. (1996) Felbamate as add-on therapy. European Neurology 36, 146–148.
Loscher, W. and Schmidt, D. (1988) Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Research 2, 145–181.
Pellock, J.M. (1999) Felbamate. Epilepsia 40(Suppl. 5), S57–62.
Pellock, J.M., Faught, E., Leppik, I.E., Shinnar, S. and Zupanc, M.L. (2006) Felbamate: consensus of current clinical experience. Epilepsy Research 71, 89–101.
Reidenberg, P., Glue, P., Banfield, C.R., Colucci, R.D., Meehan, J.W., Radwanski, E., Mojavarian, P., Lin, C.C., Nezamis, J., Guillaume, M. et al. (1995) Effects of felbamate on the pharmacokinetics of phenobarbital. Clinical Pharmacology and Therapeutics 58, 279–287.
Rho, J.M., Donevan, S.D. and Rogawski, M.A. (1994) Mechanism of action of the anticonvulsant felbamate: opposing effects on N-methyl-D-aspartate and gamma-aminobutyric acidA receptors. Annals of Neurology 35, 229–234.
Romanyshyn, L.A., Wichmann, J.K., Kucharczyk, N. and Sofia, R.D. (1993) Simultaneous determination of felbamate and three metabolites in rat and dog plasmas by high-performance liquid chromatography. Journal of Chromatography 622, 229–234.
Ruehlmann, D., Podell, M. and March, P. (2001) Treatment of partial seizures and seizure-like activity with felbamate in six dogs. Journal of Small Animal Practice 42, 403–408.
Sankar, R. and Holmes, G.L. (2004) Mechanisms of action for the commonly used antiepileptic drugs: relevance to antiepileptic drug-associated neurobehavioral adverse effects. Journal of Child Neurology 19(Suppl. 1), S6–14.
Shi, L.L., Dong, J., Ni, H., Geng, J. and Wu, T. (2011) Felbamate as an add-on therapy for refractory epilepsy. Cochrane Database Syst Rev, CD008295.
Ticku, M.K., Kamatchi, G.L. and Sofia, R.D. (1991) Effect of anticonvulsant felbamate on GABAA receptor system. Epilepsia 32, 389–391.
Wagner, M.L., Graves, N.M., Marienau, K., Holmes, G.B., Remmel, R.P. and Leppik, I.E. (1991) Discontinuation of phenytoin and carbamazepine in patients receiving felbamate. Epilepsia 32, 398–406.
White, H.S. (1999) Comparative anticonvulsant and mechanistic profile of the established and newer antiepileptic drugs. Epilepsia 40(Suppl. 5), S2–10.
White, H.S., Harmsworth, W.L., Sofia, R.D. and Wolf, H.H. (1995) Felbamate modulates the strychnine-insensitive glycine receptor. Epilepsy Research 20, 41–48.
Yang, J.T., Adusumalli, V.E., Wong, K.K., Kucharczyk, N. and Sofia, R.D. (1991) Felbamate metabolism in the rat, rabbit, and dog. Drug Metabolism and Disposition 19, 1126–1134.
Yang, J.T., Morris, M., Wong, K.K., Kucharczyk, N. and Sofia, R.D. (1992) Felbamate metabolism in pediatric and adult beagle dogs. Drug Metabolism and Disposition 20, 84–88.
19 Topiramate
Simon Platt BVM&S MRCVS Dipl. ACVIM (Neurology) Dipl. ECVN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
Topiramate (2,3:4,5-bis-O-(1-methylethylidene)ß-D-fructopyranose sulfamate) is a structurally novel antiseizure medications which is a sulfamate-substituted derivative of the monosaccharide D-fructose (Fig. 19.1; Shank et al., 2000; Guerrini and Parmeggiani, 2006).
Topiramate is an approved antiseizure medication used in adult and paediatric human patients for the treatment of focal (partial) and generalized seizures. It can be used as a single therapy agent or as adjunctive therapy (Lyseng-Williamson and Yang, 2007). It is also indicated for seizures associated with Lennox-Gastaut syndrome (see Chapter 18). There is no clinical information available about the use of this drug in dogs and cats with epilepsy.
Mechanism of Action
Topiramate produces a frequency-dependent inhibition of normal synaptic transmission and also reduces the duration of paroxysmal depolarizing shifts (PDS) evoked in vitro (Jahromi et al., 2000). The PDS is clinically correlated to interictal discharges. However, the precise mechanism by which topiramate exerts its anticonvulsant effect is unknown; however, it increases the frequency at which g-aminobutyric acid (GABA), a major inhibitory chemical of the central nervous system, activates GABAA receptors, and enhances the ability of GABA to induce a flux of chloride ions into neurons (Fig. 19.2). This suggests that topiramate potentiates the activity of this inhibitory neurotransmitter. Additionally, it has been shown to inhibit kainite/a-amino3-hydroxy-5-methylisoxazole-4-proprionic acid (AMPA)-type glutamate receptors and voltage-sensitive sodium and calcium channels (Caldwell et al., 2005; Lyseng-Williamson and Yang, 2007). Topiramate also increases potassium conductance and inhibits some carbonic anhydrase isoenzymes, although the strength of the latter property does not correlate with topiramate’s antiseizure potency.
Metabolism and Pharmacokinetics
The pharmacokinetics of topiramate in humans are dose proportional. Topiramate is rapidly absorbed, with mean peak plasma concentrations being achieved in approximately 2 h (Sachdeo et al., 2002; Lyseng-Williamson and Yang, 2007). Absorption is good and is not affected by administration with food, making it suitable for dosing outside of meal times, and protein binding is minimal (9–17%) (Perucca, 1997). Oral bioavailability is approximately 80–95% (Perucca and Bialer, 1996). Topiramate distributes into all tissues, including
Topiramate 459
the brain. Steady state in humans is achieved in approximately 4–8 days in the presence of normal renal function based on an elimination half-life of 19–25 h (Lyseng-Williamson and Yang, 2007).
From the available human data, topiramate is not metabolized extensively once absorbed, with about 70–80% of an administered dose eliminated unchanged in the urine (Lyseng-Williamson and Yang, 2007). Six trace metabolites of topiramate formed by hydroxylation, hydrolysis and glucoronidation have been identified, none of which has significant pharmacological activity (Johannessen, 1997). However, the metabolism of topiramate in people is increased during polytherapy
 NH2O S OO OO
NH2O S OO OO
O
O O
with enzyme-inducing antiseizure medications
(e.g. phenobarbitone), with the proportion of the dose metabolized increasing to 50–70% and the half-life reduced by 50% (Bialer et al., 2004; Perucca, 2006). Clearance of topiramate is also reduced up to 54% in patients with renal impairment, necessitating dosage adjustments.
In healthy beagle dogs administered 40 mg/kg of topiramate, peak plasma concentration occurs between 0.6 and 3.8 h (Streeter et al., 1995). The absolute bioavailability of an oral dose of topiramate was estimated to be in the range of 27 to 59%, depending on the formulation. Oral plasma clearance and terminal half-life values were found to be 2.4–3.6 ml/min/kg and 2.6–3.7 h following a single oral administration, and 3.0–4.2 ml/min/kg and 2.0–3.8 h after multiple doses. Plasma protein binding is as low as it is in humans (8–13%). There was no evident accumulation and no autoinduction or inhibition of enzymes that metabolize topiramate resulting from multiple dosing (Streeter et al., 1995). Topiramate is not extensively metabolized in dogs and is primarily eliminated unchanged in the urine. However, significant biliary excretion is present following topiramate administration in dogs (Caldwell et al., 2005). There is no available information on the pharmacokinetics of this drug in cats.
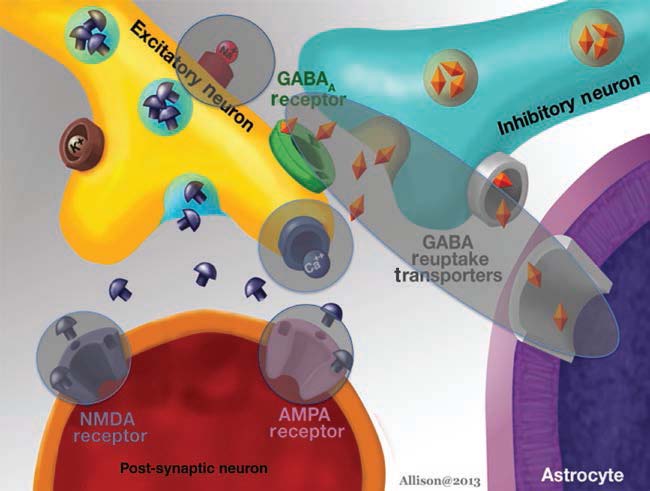
Fig. 19.2. The sites of action of topiramate at the neuronal synapse are highlighted.
Pharmacokinetic Interactions and Adverse Reactions
Topiramate has a relatively low potential for clinically relevant interactions with other medications (Johannessen, 1997; Bialer et al., 2004). It is not an inhibitor in vitro of the cytochrome P450 (CYP) isoenzymes CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2D6, CYP2E1 and CYP3A4/5, and inhibits CYP2C19 only at high plasma concentrations (equivalent to topiramate dosages 5- to 15-fold higher than the recommended dosages (Sachdeo et al., 2002). Topiramate has only a weak potential to induce other CYP isozymes. It is susceptible to drug interactions that result from induction of its metabolism during polytherapy with enzyme-inducing antiepileptic medications. However, when topiramate is coadministered with phenobarbital in humans, there is limited change in the plasma concentrations of phenobarbitone or topiramate (Lyseng-Williamson and Yang, 2007). Unlike felbamate, the use of topiramate with phenobarbital is synergistic, albeit through poorly understood mechanisms (Czuczwar and Przesmycki, 2001).
The most commonly observed CNS and peripheral nervous system related side-effects in humans associated with the use of topiramate are somnolence, dizziness, ataxia, vertigo and speech disorders, seen in approximately 30% of patients (Shorvon, 1996). Other adverse events associated with this medication include cognitive and psychiatric events, metabolic acidosis, renal calculi, loss of appetite and weight loss. The metabolic acidosis and renal calculi are potentially linked to the carbonic anhydrase inhibitory effects of topiramate. The tolerability of topiramate improves with slower rates of titration. In a prospective post-marketing human study, 41.4% of patients with focal seizures who received topiramate experienced cognitive adverse events (Tatum et al., 2001). The most commonly reported cognitive effect is psychomotor slowing. Overall up to 53% of people taking this medication experienced some sort of side effects, which seem to become more prevalent with chronic therapy (Bootsma et al., 2004; Al Ajlouni et al., 2005). This issue prevents the more common use of this medication in the long term.
No adverse reactions were reported in healthy beagle dogs administered 10–150 mg/ kg daily oral doses for 15 days (Streeter et al., 1995). Dosing for 3 and 12 months was not associated with overt indicators of toxicity. Dogs treated with up to 400 mg/kg experienced ataxia, decreased motor activity, tremors and clonic convulsions.
Dosing and Monitoring Recommendations
Topiramate is generally administered in divided doses and may be taken with or without food. The medication comes in sprinkle capsules, which may be opened and the contents sprinkled on soft food. This mode of administration may be useful in dogs and cats.
The dose of topiramate should be reduced by 50% in patients with renal impairment; however, dosage reductions are not necessary in patients with hepatic impairment (Garnett, 2000).
In epileptic human patients, plasma topiramate concentrations at steady state have generally been found to increase linearly with increasing daily dosage within the clinically used dose range (Sachdeo et al., 1996). Although plasma topiramate concentrations have been measured in many trials, no consistent relationship with clinical response has been documented in humans. Topiramate concentrations are generally higher in patients with adverse effects (particularly anorexia, weight loss, impaired concentration and anxiety) than in those without adverse effects. However, there is no indication that monitoring plasma topiramate levels would improve clinical outcome in terms of efficacy or safety.
There are currently no dosing or monitoring recommendations published on the use of topiramate in the dog or cat.
Topiramate 461
Efficacy
Topiramate shows relatively broad-spectrum antiseizure properties in experimental animal studies (Perucca, 1997). Models in which it has shown seizure-protecting activity include the maximal electroshock (MES) test in mice and rats, the genetically seizure-prone DBA/2 mouse, the spontaneously epileptic rat and the amygdala kindled rat (Shank et al., 1994; Wauquier and Zhou, 1996; Perucca, 1997). In the MES test, the effect of topiramate was comparable to that of phenobarbital and was more favourable when considering toxic side-effects at the effective dose. Topiramate is inactive or weakly active in a number of chemically induced seizure models in rodents, including the clonic or tonic seizures induced by picrotoxin, pentylenetetrazol, bicuculline and strychnine (Wauquier and Zhou, 1996). Overall, these experiments suggest that its main action involves a block in seizure spread rather than elevation in seizure threshold (Shank et al., 2000).
Topiramate has been proven an effective monotherapy and adjunctive agent for reducing the number of both focal and generalized tonic-clonic seizures experienced by people with epilepsy (Lyseng-Williamson and Yang, 2007). Various human studies have shown that the proportion of seizure-free patients on this medication ranges from 4 to 83%, depending on the age of patient, duration of treatment and type of seizure (Reife et al., 2000; Al Ajlouni et al., 2005; Arroyo et al., 2005; Guerrini et al., 2005). Across all epilepsy types, reductions in seizure frequency were observed in 44–64% of patients with the mean percentage reduction of seizures being 58% for secondarily generalized seizures and 57% for simple focal seizures (Perucca and Bialer, 1996).
Nasogastric topiramate solution administered twice daily has been evaluated for the treatment of refractory status epilepticus in humans (Towne et al., 2003). The treatment has been shown to be effective within 24 h. At the current time though, a parenteral solution is not available.
There are no data available on the potential efficacy of topiramate in dogs or cats.
Summary Recommendations
• Topiramate may be a useful medication
to consider for the treatment of all refractory seizure types in dogs and cats.
• Clinical information on the use of this
medication in dog and cats is lacking.
• Routine monitoring of plasma topiramate
concentrations cannot be recommended and the need for dosage adjustments should be determined by clinical observation.
• The incidence of side effects seen with its
use in humans means that it is currently a fourth or fifth line medication.
References
Al Ajlouni, S., Shorman, A. and Daoud, A.S. (2005) The efficacy and side effects of topiramate on refractory epilepsy in infants and young children: a multi-center clinical trial. Seizure 14, 459–463.
Arroyo, S., Dodson, W.E., Privitera, M.D., Glauser, T.A., Naritoku, D.K., Dlugos, D.J., Wang, S., Schwabe, S.K. and Twyman, R.E. (2005) Randomized dose-controlled study of topiramate as first-line therapy in epilepsy. Acta Neurologica Scandanavica 112, 214–222.
Bialer, M., Doose, D.R., Murthy, B., Curtin, C., Wang, S.S., Twyman, R.E. and Schwabe, S. (2004) Pharmacokinetic interactions of topiramate. Clinical Pharmacokinetics 43, 763–780.
Bootsma, H.P., Coolen, F., Aldenkamp, A.P., Arends, J., Diepman, L., Hulsman, J., Lambrechts, D., Leenen, L., Majoie, M., Schellekens, A. and De Krom, M. (2004) Topiramate in clinical practice: long-term experience in patients with refractory epilepsy referred to a tertiary epilepsy center. Epilepsy and Behavior 5, 380–387.
Caldwell, G.W., Wu, W.N., Masucci, J.A., Mckown, L.A., Gauthier, D., Jones, W.J., Leo, G.C. and Maryanoff, B.E. (2005) Metabolism and excretion of the antiepileptic/antimigraine drug, Topiramate in animals and humans. European Journal of Drug Metabolism and Pharmacokinetics 30, 151–164.
Czuczwar, S.J. and Przesmycki, K. (2001) Felbamate, gabapentin and topiramate as adjuvant antiepileptic drugs in experimental models of epilepsy. Polish Journal of Pharmacology 53, 65–68.
Garnett, W.R. (2000) Clinical pharmacology of topiramate: a review. Epilepsia 41(Suppl. 1), S61–65.
Guerrini, R. and Parmeggiani, L. (2006) Topiramate and its clinical applications in epilepsy. Expert Opinion on Pharmacotherapy 7, 811–823.
Guerrini, R., Carpay, J., Groselj, J., Van Oene, J., Schreiner, A., Lahaye, M. and Schwalen, S. (2005) Topiramate monotherapy as broad-spectrum antiepileptic drug in a naturalistic clinical setting. Seizure 14, 371–380.
Jahromi, S.S., Pelletier, M.R., Mcdonald, P.J., Khosravani, H. and Carlen, P.L. (2000) Antiepileptic efficacy of topiramate: assessment in two in vitro seizure models. Brain Research 872, 20–28.
Johannessen, S.I. (1997) Pharmacokinetics and interaction profile of topiramate: review and comparison with other newer antiepileptic drugs. Epilepsia 38(Suppl. 1), S18–23.
Lyseng-Williamson, K.A. and Yang, L.P. (2007) Topiramate: a review of its use in the treatment of epilepsy. Drugs 67, 2231–2256.
Perucca, E. (1997) A pharmacological and clinical review on topiramate, a new antiepileptic drug. Pharmacological Research 35, 241–256.
Perucca, E. (2006) Clinical pharmacokinetics of new-generation antiepileptic drugs at the extremes of age. Clinical Pharmacokinetics 45, 351–363.
Perucca, E. and Bialer, M. (1996) The clinical pharmacokinetics of the newer antiepileptic drugs. Focus on topiramate, zonisamide and tiagabine. Clinical Pharmacokinetics 31, 29–46.
Reife, R., Pledger, G. and Wu, S.C. (2000) Topiramate as add-on therapy: pooled analysis of randomized controlled trials in adults. Epilepsia 41(Suppl. 1), S66–71.
Sachdeo, R.C., Sachdeo, S.K., Walker, S.A., Kramer, L.D., Nayak, R.K. and Doose, D.R. (1996) Steady-state pharmacokinetics of topiramate and carbamazepine in patients with epilepsy during monotherapy and concomitant therapy. Epilepsia 37, 774–780.
Sachdeo, R.C., Sachdeo, S.K., Levy, R.H., Streeter, A.J., Bishop, F.E., Kunze, K.L., Mather, G.G., Roskos, L.K., Shen, D.D., Thummel, K.E., Trager, W.F., Curtin, C.R., Doose, D.R., Gisclon, L.G. and Bialer, M. (2002) Topiramate and phenytoin pharmacokinetics during repetitive monotherapy and combination therapy to epileptic patients. Epilepsia 43, 691–696.
Shank, R.P., Gardocki, J.F., Vaught, J.L., Davis, C.B., Schupsky, J.J., Raffa, R.B., Dodgson, S.J., Nortey, S.O. and Maryanoff, B.E. (1994) Topiramate: preclinical evaluation of structurally novel anticonvulsant. Epilepsia 35, 450–460.
Shank, R.P., Gardocki, J.F., Streeter, A.J. and Maryanoff, B.E. (2000) An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia 41(Suppl. 1), S3–9.
Shorvon, S.D. (1996) Safety of topiramate: adverse events and relationships to dosing. Epilepsia 37(Suppl. 2), S18–S22.
Streeter, A.J., Stahle, P.L., Holland, M.L., Pritchard, J.F. and Takacs, A.R. (1995) Pharmacokinetics and bioavailability of topiramate in the beagle dog. Drug Metabolism and Disposition 23, 90–93.
Tatum, W.O.T., French, J.A., Faught, E., Morris, G.L. 3rd, Liporace, J., Kanner, A., Goff, S.L., Winters, L. and Fix, A. (2001) Postmarketing experience with topiramate and cognition. Epilepsia 42, 1134–1140.
Towne, A.R., Garnett, L.K., Waterhouse, E.J., Morton, L.D. and Delorenzo, R.J. (2003) The use of topiramate in refractory status epilepticus. Neurology 60, 332–334.
Wauquier, A. and Zhou, S. (1996) Topiramate: a potent anticonvulsant in the amygdala-kindled rat. Epilepsy Research 24, 73–77.
20 The New Additions: Lacosamide,
Brivaracetam and Rufinamide
Simon Platt BVM&S MRCVS Dipl. ACVIM (Neurology) Dipl. ECVN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
Lacosamide
Lacosamide (LCM) (SPM 927, formerly harkoseride), the R-enantiomer of 2-acetamidoN-benzyl-3-methoxypropionamide, is a series of functionalized amino acids, with antiseizure and antinociceptive properties (Fig 20.1) (Beyreuther et al., 2007). In November 2007, a new drug application was filed with the FDA in the USA for use of LCM as adjunctive therapy in the treatment of focal-onset seizures in adult humans with epilepsy. Three formulations are produced: tablets, syrup and an intravenous injection (Biton et al., 2008; Cawello et al., 2012a, b). LCM was approved in Europe on 3 September 2008 as adjunctive therapy in the treatment of focal-onset seizures, with or without secondary generalization (Halford and Lapointe, 2009).
Mechanism of Action
LCM causes a general decrease in neuronal discharge frequency and synaptic excitability in laboratory testing. Since these actions are not mediated at major excitatory (AMPA/ NMDA) or inhibitory (GABA) postsynaptic receptors, the mechanism of action was initially unknown (Errington et al., 2006; Halford and Lapointe, 2009). Recently two different probable mechanisms of action have been discovered:
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
whether the effect on this protein is stimulatory or inhibitory, but as a consequence LCM appears to attenuate the effects of neurotrophic factors on axon outgrowth (Beyreuther et al., 2007). The CRMP family of proteins is implicated in developmental processes of the nervous system, since most of the five CRMP proteins are highly expressed during early development and mainly in the central nervous system. On an in vitro level, CRMP-2 has been shown to be involved in neuronal differentiation, polarization and axonal outgrowth induced by neurotrophic factors such as brain-derived neurotrophic factor (BDNF) or neurotrophin-3 (NT-3) (Halford and Lapointe, 2009). Ongoing experiments aim at showing that LCM can interfere with these NT-induced processes, thereby potentially attenuating the
HN OO
NH O

development and/or progression of epilepsy and/or neuropathic pain. A recent study showing that CRMP-2 is deregulated in human brain samples from treatment-resistant epilepsy patients but not from control patients makes this hypothesis even more plausible (Czech et al., 2004). Additional in vitro experiments suggest that CRMP-2 is involved in neuronal protection from excitotoxicity and apoptosis. LCM has shown strong neuroprotective and anti-apoptotic effects following glutamate-induced excitotoxicity in hippocampal slices. Moreover, neuroprotective effects of LCM were also observed in in vivo animal models, for example, following status epilepticus.
Metabolism and Pharmacokinetics
In humans, LCM is rapidly and almost completely absorbed from the gastrointestinal tract, with an oral bioavailability of approximately 100% (Halford and Lapointe, 2009). LCM is absorbed within 4 h and food does not affect its bioavailability. The intravenous formulation of LCM has been studied in short-term replacement therapy and appeared to be
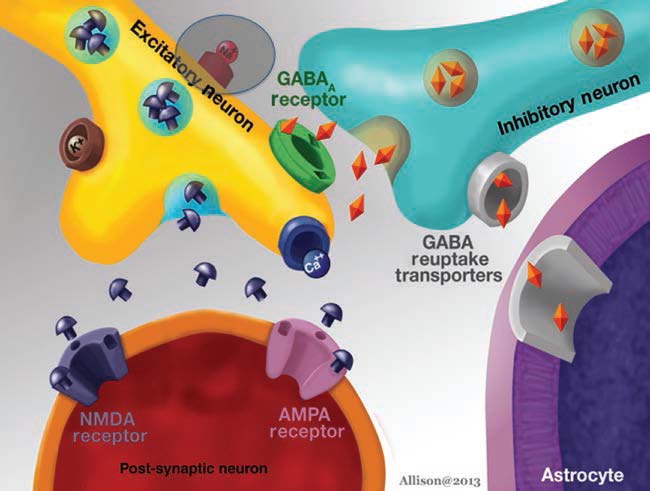
Fig. 20.2. Neuronal receptor targets for lacosamide and rufinamide. Both lacosamide and rufinamide have the same site of action.
Lacosamide, Brivaracetam and Rufinamide
Resting membrane
Open potential Resting statestate LACOSAMIDE hyperactivity
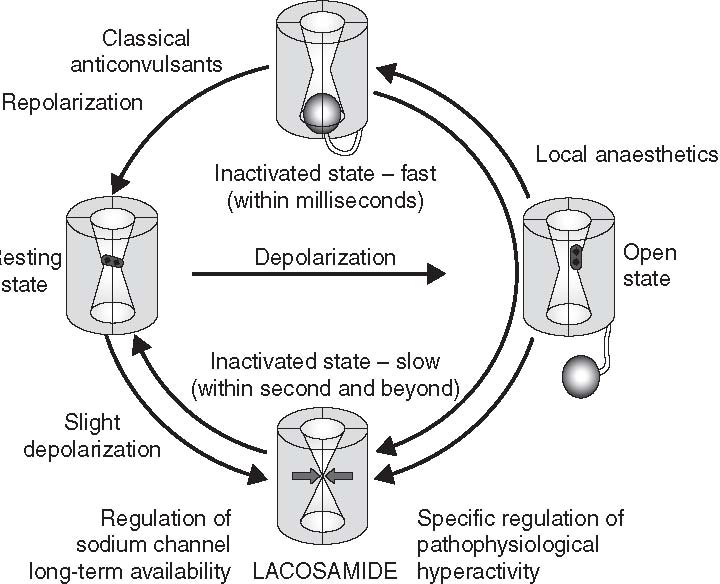
Fig. 20.3. Physiology of voltage-gated sodium channels. Depending on the membrane potential and the neuronal activity, voltage-gated sodium channels are in different states. At the resting potential sodium channels are closed and can be opened by depolarization of the membrane potential allowing the flux of sodium ions into the cell. Within a few milliseconds the channels close from the inside of the neuron and go into the fast inactivated state from which they cannot be activated. When the membrane potential returns to its baseline the sodium channel goes back to its resting state. Under conditions of slight prolonged depolarization and repetitive neuronal activity the sodium channel can go into the slow inactivated state by closing the pore from the inside. This process happens on a second-to-minute time scale. Drugs can either block the open channel (e.g. local anaesthetics), or enhance fast inactivation (classical anticonvulsants) or enhance slow inactivation (lacosamide). (Beyreuther et al., 2007.)
safe and well tolerated (Biton et al., 2008; Cawello et al., 2012b; Fountain et al., 2013). LCM undergoes minimal first-pass metabolism and is eliminated by renal excretion (40%) and biotransformation. The primary metabolite from demethylation (CYP2C19) has no activity and is eliminated by the kidney (30% of the dose). LCM has a half-life of 13 h, displays a first-order pharmacokinetic elimination profile and is administered twice daily. Pharmacokinetic studies showed a predictable volume of distribution and elimination rate, and no clinically significant differences between the pharmacokinetics of LCM in young and elderly subjects were observed (Halford and Lapointe, 2009). Protein-binding is minimal (less than 15%) and plasma concentrations are consequently linear and dose proportional; steady state is achieved in 3 days. Therapeutic concentrations with LCM have not been established in humans or animals; drug-level monitoring is not necessary. Moderate hepatic and renal impairment have both been shown to increase systemic drug exposure up to approximately 40% (Halford and Lapointe, 2009). Titration, therefore, should be carried out with caution in these groups of patients.
Following administration of 3 mg/kg lacosamide orally to hound-cross dogs, the total oral clearance of lacosamide was determined to be 0.0958 ± 0.0398 l/h/kg (Martinez et al., 2012). The apparent oral volume of distribution (Vd) of lacosamide was 0.402 ± 0.122 l/kg. The serum concentrations of lacosamide appeared to decline with a mean serum elimination half-life of 3.58 ± 1.40 h. The time to maximum concentration (Tmax) was 2.33 ± 1.44 h with a concentration maximum (Cmax) of 8.06 ±
5.46 mg/ml (Martinez et al., 2012). The area under the curve, representing the total amount of drug exposure in the serum over

| Effects on | Enhanced | Changes in | Effects on |
|---|---|---|---|
| neuronal | axonal | gene | myelination |
| sprouting | outgrowth | expression | |
Fig. 20.4. Schema showing CRMP-2-mediated transduction of neurotrophic signals to neuronal response and the possible interaction of lacosamide. Neurotrophins like NT-3 and BDNF activate their receptors in the plasma membrane, triggering a transduction cascade, which regulates the activity of intracellular protein kinases (e.g., PI3 kinase or GSK-3b ) finally resulting in increased levels of active CRMP-2. Active, nonphosphorylated CRMP-2 has been shown to enhance axonal outgrowth and might also be involved in the induction of other cellular responses. Interaction site of lacosamide is indicated. (Beyreuther et al., 2007.)
time, was 43.4 ± 29.1 mg/h/ml. At the time of writing, no similar information could be found on the metabolism and pharmacokinetics of this drug in cats.
Pharmacokinetic Interactions and Adverse Reactions
LCM displays a favourable interaction profile with currently prescribed anti-epileptic medications and other commonly used medications. A clinical pharmacokinetic study with healthy human subjects demonstrated that concomitant administration of LCM with the commonly prescribed antiepileptic medications (AEMs) does not significantly affect its rate and its extent of absorption or that of the antiepileptic medication. LCM had no relevant influence on plasma concentrations of concomitant antiepileptic medications (including gabapentin, topiramate, levetiracetam and phenobarbitone) for human patients with focal seizures, with or without secondary generalization. In population pharmacokinetic studies, the blood level of LCM is decreased by approximately 15–20% by enzyme-inducing anti-epileptic medications (Curia et al., 2009). No food–drug or drug–drug interactions with other marketed agents (e.g. omeprazole) that could be affected by the CYP2C19 pathway or protein binding at therapeutic levels have been identified. Overall, the repeated dose toxicity studies have demonstrated that LCM is well tolerated in rats and dogs after either
Lacosamide, Brivaracetam and Rufinamide
intravenous or oral administration. Similarly, repeated-dose oral administration of LCM to mice was not associated with significant toxicity. The dog was slightly more sensitive to LCM exposure than were rodents. All treatment-related effects observed in rats and dogs were completely reversible within a 4-week recovery period. Clinical signs observed in the chronic toxicity studies in mice, rats and dogs were dose dependent and included neurological signs such as ataxia, reduced motor function, tremor or convulsions at high doses. In most cases, these signs can be attributed to exaggerated pharmacodynamic effects of LCM and were considered dose-limiting. Similar CNS-related pharmacodynamic effects were also reported from preclinical studies with other AEMs like pregabalin (Beyreuther et al., 2007). Based on animal studies to date (mostly in rats and dogs), LCM has no effect on the autonomic nervous system and does not have clinically significant adverse effects on the gastrointestinal, renal or respiratory systems.
Two studies have shown that LCM solution for infusion is safe and well tolerated when given as an intravenous infusion over the time periods of 10, 15, 30 and 60 min in doses ranging from 200 mg to 600 mg/day (Biton et al., 2008; Krauss et al., 2010). Few adverse events were reported during these intravenous studies, and the safety profile for LCM injection was comparable to oral LCM tablets, based on analyses of adverse events, ECGs, vital signs and laboratory parameters (Biton et al., 2008; Cawello et al., 2012b, 2013).
Based on several human trials, LCM is well tolerated (Krauss et al., 2012; Fountain et al., 2013). Most documented adverse events are mild to moderate and commonly observed during the titration phase. The most commonly experienced effects are associated with the CNS and gastrointestinal tract. Side-effects in people which have led to medication dis-continuation include dizziness (up to 53% of patients experience and is dose-dependent), nausea, ataxia, vomiting and nystagmus (Beyreuther et al., 2007; Curia et al., 2009). Somnolence is very uncommon with this medication. LCM does not cause weight changes; neither does it change patterns in haematology, blood chemistry and vital signs.
Dosing and Monitoring Recommendations
Current literature indicates that lacosamide should be used cautiously in human patients with severe cardiac disease and in those who are taking medications known to induce PR interval prolongation (Chinnasami et al., 2013). People with underlying cardiac abnormalities should have an ECG performed before initiating lacosamide and after steady-state is achieved. Lacosamide should be titrated slowly at withdrawal to minimize its potential to increase seizure frequency (Shaibani et al., 2009).
HPLC analysis of LCM has been successfully reported in dogs (Martinez et al., 2012). A serum reference range for people has been established as 3–14 mg/l (Krasowski, 2010). No dosing recommendations have currently been published for the use of LCM in dogs although it is likely to require three times daily dosing based on its serum half-life.
Efficacy
LCM has a unique profile when studied across a range of animal models of epilepsy, demonstrating an antiseizure effect similar to many of the newer antiseizure medications. It is effective in the maximal electroshock model and elevates the seizure threshold in the pentylenetratazol seizure test (Stohr et al., 2007). A kindling seizure model has been used to evaluate whether LCM affects kindling-induced epileptogenesis. The rats were treated with different doses of LCM (3, 10, or 30 mg/kg/ day) over 22–23 days during amygdala kindling. Daily administration of LCM during kindling acquisition produced a dose-dependent effect on kindling development. Although the drug was inactive at 3 mg/kg/day, significant retardation of kindling was observed at 10 mg/kg/day, at which the average number of stimulations to reach kindling criteria was increased by >90%. These data demonstrate that LCM, in addition to exerting anticonvulsant activity, has the potential to retard kindling-induced epileptogenesis (Brandt et al., 2006).
LCM also decreases self-sustaining status epilepticus (SSSE) in rats, inhibits NMDA-induced seizures in mice and is able to completely block 4-aminopyridine-induced seizures in vitro (Lees et al., 2006; Stohr et al., 2007). Spike frequency and cumulative time spent in seizures significantly decreased; only separate spikes were recorded for 12 h after induction of SSSE (Wasterlain et al., 2011). In the control group, three of the six animals died, whereas, in the treatment group, all survived. Histological examination of brain sections (dorsal hippo-campus) collected 72 h after status epilepticus revealed significantly less damage in LCM-treated rats compared with control animals, suggesting LCM may be neuroprotective (Beyreuther et al., 2007).
As an add-on medication, LCM has been shown to significantly reduce seizure activity in human patients with focal epilepsy (Halford and Lapointe, 2009). The median percentage reduction in seizure frequency when LCM was used as an adjunctive therapy ranged from 26 to 40% (Ben-Menachem et al., 2007; Curia et al., 2009; Chung et al., 2010). Up to 8% of patients in the adjunctive studies were completely seizure free and long-term response has been demonstrated (Yorns et al., 2012). The potential value of LCM as a first-line therapy and in the treatment of generalized epilepsies is unknown. Recent work has demonstrated the benefit of this medication for the treatment of human status epilepticus (Hofler et al., 2011; Kellinghaus et al., 2011; Hofler and Trinka, 2013). Status epilepticus cessation was observed in 42 human patients (88%) in one study (Hofler et al., 2011). Success in treating humans with SE LCM as a first or second drug was 100% (8 of 8), as a third drug 81% (11 of 15) and as a fourth or later drug 75% (6 of 8) (Hofler et al., 2011; Kellinghaus et al., 2011). There were no side effects observed except for pruritus and skin rash in two patients in this study. Such data support use of IV LCM as a potential alternative to standard AEMs for acute treatment of seizures although randomized controlled studies are needed.
Summary Recommendations
• LCM has a novel dual mechanism of
action, that is, selective enhancement of
sodium channel slow inactivation and modulation of CRMP-2 activity.
• LCM demonstrates anti-epileptic effectiveness for generalized and focal seizures as well as for status epilepticus.
• Its use in dogs and cats should proceed
with caution until the short- and long-term side effects have been established.
Brivaracetam
Brivaracetam ((2S)-2-((4R)-2-oxo-4-propylpyrrolidinyl) butanamide) is a novel member of the piracetam family of anticonvulsants (Fig. 20.5). Brivaracetam (BRV) is a next-generation drug to levetiracetam (LEV) and has recently been attributed with potentially superior antiseizure activities based on in vitro drug screening and animal tests (Malykh and Sadaie, 2010). The higher potency of BRV is partially consistent with recent clinical results in humans. This medication is available in the USA and in Europe.
Mechanism of Action
The pharmacology of the piracetam-related drugs has been less explored than the clinical applications of these drugs and remains to be elucidated. In general, these compounds interact with target receptors in the brain and modulate the excitatory and/or inhibitory processes of neurotransmitters, neurohormones and/or post-synaptic signals (Malykh and Sadaie, 2010). The similarity of its chemical structure to a cyclic derivative of GABA suggests that piracetam compounds probably have a GABA-mimetic action.
O
NH2 N
O

Lacosamide, Brivaracetam and Rufinamide
Unlike some piracetam compounds, levetiracetam (LEV), the prototype anticonvulsant of this group of drugs, apparently inhibits neuronal Ca2+ ion channels that are possibly important to its antiseizure effect (Fig. 20.6) (Pisani et al., 2004; Carunchio et al., 2007). In a different experimental setting using a seizure model in mice, it was later demonstrated to bind to synaptic vesicle 2A (SV2A) protein in brain membranes and fibroblasts (Lynch et al., 2004). The data correlated with the clinical application of LEV as an antiseizure medication. BRV, the newer-generation chemical entity after LEV, binds to SV2A with a ten-fold higher affinity and has been evaluated clinically for its anti-epileptic properties in humans (Tai and Truong, 2007). BRV also inhibits voltage-dependent Na+ currents at therapeutically relevant concentrations (Zona et al., 2010).
Metabolism and Pharmacokinetics
In human patients, BRV appears to be well absorbed with linear pharmacokinetics, and with a median tmax between 1 and 2 h with low coefficients of variation of Cmax (<25%).
Metabolism takes place primarily via hydrolysis and secondarily through hydroxylation mediated by cytochrome (CYP) 2C19. The main metabolites (acid, hydroxyl and hydroxyacid) are not pharmacologically active (French et al., 2010).
The half-life has been calculated to be approximately 8 h in humans. There appears to be no clear evidence of CYP3A4 induction, suggesting a low potential for interaction with medications that are substrates for this enzyme. Urinary recovery of the parent drug over one dosing interval is between 5 and 8%, with between 33 and 49% additionally recovered as two metabolites. Renal clearance of the parent drug was approximately 3 ml/min. Considering that binding to plasma proteins is low (<20%), this suggests that substantial tubular reabsorption must occur (Rolan et al., 2008). The observed limited renal excretion of unchanged BRV (5–8%) together with the presence of significant levels of metabolites in the urine indicates that the compound is predominantly cleared by metabolism. The increase in apparent total body clearance was only modest, indicating that more frequent dosing is not warranted at higher doses of BRV.
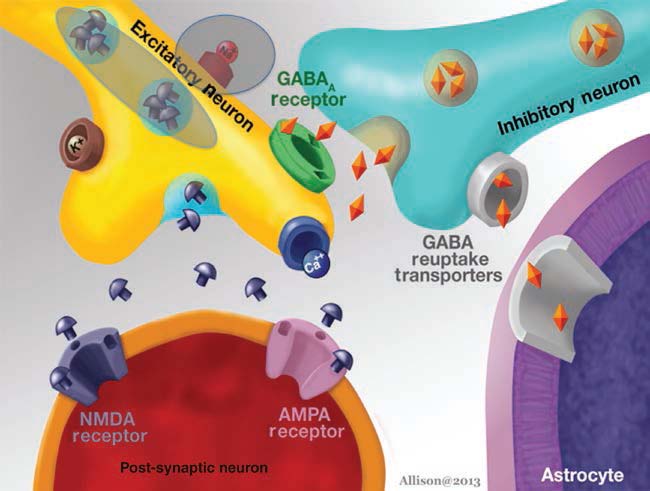
Pharmacokinetic Interactions and Adverse Reactions
All adverse events associated with the administration of BRV have been reported to be either mild or moderate and almost all have been related to the central nervous system (CNS) in humans (Rolan et al., 2008). The most frequently reported adverse events include dizziness (33–89%) and euphoria (11–44%), and these occur in a dose-related manner. At higher doses, adverse events were most likely to be reported on the first study day with the frequency being similar across all treatment groups (including placebo) from the second day onwards, suggesting rapid tolerance to the adverse effects (Rolan et al., 2008).
Dosing and Monitoring Recommendations
At present, there are no consistent recommendations for the dosing and monitoring of BRV in humans or dogs and cats. The reader is referred to the chapter on LEV (Chapter 16) as the monitoring recommendations for that medication are likely to apply in the future to BRV.
Efficacy
Preclinical testing demonstrated that BRV induced a more potent and complete suppression of seizures and kindling acquisition than did LEV in various in vitro and in vivo models of epilepsy (Matagne et al., 2008). The antiseizure activities observed with BRV in animal models that are thought to mimic partial-onset (kindled animals) and generalized (audiogenic seizure-susceptible mice) epilepsy in humans were superior to those observed with LEV. Pretreatment with BRV during the corneal kindling process in mice revealed a potent and persistent ability to inhibit kindling development, which was superior to that of LEV (Matagne et al., 2008). BRV also showed a wide therapeutic index in kindled animals, similar to that of LEV. Taken together, these results demonstrate that BRV possesses properties superior to LEV as an anti-epileptic and anti-epileptogenic agent in various experimental models of epilepsy.
BRV has been shown to be effective in humans with focal onset seizure activity with or without secondary generalization and displays a clear dose-response relationship (dose range 5–50 mg/day) for all the different efficacy analyses (reduction of weekly frequency over that seen with placebo; percentage reduction from baseline in weekly seizure frequency; ³50% responder rate) (French et al., 2010). A trial looking at the use of BRV for uncontrolled focal seizures in humans found that 50% responder rates were 17.3% for placebo compared with 35.8% for BRV at 50 mg/ day (p = 0.038) and 30.8% for BRV at 150 mg/ day (p = 0.114) (Van Paesschen et al., 2013). Nine patients were free from focal-onset seizures during the 10-week treatment period (five patients (9.4%) in the BRV 50 mg/day group and three (5.8%) in the BRV 150 mg/ day group compared with one patient (1.9%) in the placebo group). Adverse events reported during the study period were mostly mild-tomoderate with similar incidence across treatment groups (BRV 50 mg/day 36/53, 67.9%; BRV 150 mg/day 35/52, 67.3%; placebo 37/52, 71.2%) (Van Paesschen et al., 2013). The most frequently reported adverse events in BRV groups were headache, fatigue, nasopharyngitis, nausea, somnolence and dizziness, although nausea had a higher incidence in the placebo group (Van Paesschen et al., 2013).
No clinical trials have been published on the use of BRV in dogs or cats.
Summary Recommendations
• BRV may be a more potent antiseizure
medication than LEV but at this time there are not enough data present to justify its efficacious and safe use in dogs or cats.
Rufinamide
Rufinamide is a novel anticonvulsant (triazole derivative) whose mechanism of action is to prolong the inactivated state of voltage-gated
Lacosamide, Brivaracetam and Rufinamide
sodium channels (Fig. 20.7) (Krasowski, 2010). Rufinamide was approved for use in Europe in January 2007 and by the FDA in the USA in December 2008 for Lennox-Gastaut syndrome. Lennox-Gastaut syndrome, also known as Lennox syndrome, is a difficult-to-treat form of childhood-onset epilepsy that most often appears between the second and sixth year of life, and is characterized by frequent seizures and different seizure types; it is often accompanied by developmental delay and psychological and behavioural problems.
Mechanism of Action
The proposed mechanism of action is limitation of sodium-dependent action potentials (Fig. 20.2). This results in possible membrane stabilizing effects. Radio-ligand binding studies showed no interactions with monoamine, acetylcholine, histamine, glycine, AMPA (alpha-amino-3-hydroxy-5-methyl4-isoxazolepropionic acid)/kainate, NMDA (N-methyl-D-aspartic acid), GABA (gammaaminobutyric acid) receptors or systems (Bialer et al., 1999).
Metabolism and Pharmacokinetics
In humans, oral rufinamide is 70% absorbed in the fed state. In preliminary studies, food increases absorption (area under the curve (AUC) 81.7 versus 57.2 mcg h/ml), shortens T (6 versus 8 h) and increases C (4.29
maxmax
versus 2.19 mcg h/ml). Peak plasma concentration is reached in 5–6 h, independent of dose. This increased absorption should prompt consideration of dosing with food, if the slightly increased fluctuation in blood level is clinically
F N
N
N
F NH2 O

Fig. 20.7. Molecular structure of rufinamide.
acceptable. Of note, the highest fluctuations in drug levels are seen if one dose is given with food and another while fasting. Rufinamide is 34% protein bound. It is extensively metabolized by non-CYP450 systems (carboxylesterases in the liver) with only 2% unchanged in urine and faeces. The main metabolite, CGP47292, is formed by hydrolysis of the carboxylamide group. The elimination half-life is 8–12 h.
In dogs, the pharmacokinetics of rufinamide were calculated following administration of a single mean oral dose of 20.0 mg/kg (range 18.6–20.8 mg/kg) (Wright et al., 2012). No adverse effects were observed. The mean terminal half-life was 9.86 ± 4.77 h, comparable to that observed in humans and is expected to be similarly adequate to allow twice-daily dosing in clinical practice. The variability in calculated half-life values among dogs, however, suggests that every 8-h dosing may be required in some patients. The mean maximum plasma concentration achieved after a single dose of 20 mg/kg was 19.6 ± 5.8 mg/ml. While an estimated steady-state concentration cannot be extrapolated from this limited study, it is guaranteed to be higher than
19.6 mg/ml. This value exceeds the desired therapeutic level of 15 mg/ml in the treatment of human epilepsy, and while the therapeutic level in dogs may differ, this finding suggests that rufinamide will reach an effective steady state to enable its use clinically in canine patients. Studies are required to determine how concurrent liver or kidney disease might alter the pharmacokinetics of rufinamide.
Pharmacokinetic Interactions and Adverse Reactions
Rufinamide is not metabolized by the cytochrome P450 system, limiting its potential for drug–drug interactions; however, several drug–drug interactions have been reported. Phenobarbitone, phenytoin and primidone increase rufinamide clearance by 25–46% (Willmore, 2000). Rufinamide does not affect trough concentrations of phenytoin, phenobarbitol, primidone, topiramate and clonazepam (Bialer et al., 1999).
Reported adverse effects are mild to moderate and do not lead to discontinuation in humans. These include fatigue (20%, placebo 4%), somnolence (24%, placebo 13%), tremor (12%, placebo 0%), dizziness (8%, placebo 0%) and vomiting (22%, placebo 6%), headache (varies from below placebo to up to 8% above placebo), diplopia (frequency not reported), vomiting (22%, placebo 6.3%) and diarrhoea (frequency not reported) (Pålhagen et al., 2001; Glauser et al., 2008). In a metaanalysis, discontinuation rate because of adverse effects was 8.1% in the rufinamide group and 4.3% in the placebo group (Cheng-Hakimian et al., 2006). Death was reported in 0.2% of the rufinamide group and 0.6% of the placebo group. Laboratory parameters, including ECG, were not significantly altered in adults or children. Serious adverse effects were reported in 6.3% of rufinamide-treated and 3.9% of placebo-treated patients (Cheng-Hakimian et al., 2006).
Dosing and Monitoring Recommendations
In humans, rufinamide should be given with food and daily doses should be divided equally for twice-daily administration. While rufinamide has shown efficacy in people at a dose as low as 10 mg/kg/day (Brodie et al., 2009), the recommended dose for use in the treatment of refractory epilepsy is 45 mg/kg/ day divided into two doses given 12 h apart. To decrease the likelihood of adverse effects, the dose of rufinamide is gradually increased over approximately 1 week beginning at 10 mg/ kg/day and resulting in the target dose of 45 mg/kg/day. At a dosage of 45 mg/kg/day, the typical plasma steady-state concentration achieved in humans is approximately 15 mg/ml, which represents the average therapeutic plasma concentration of rufinamide.
Rufinamide requires tapered discontinuation at a suggested rate of 25% dose reduction every 2 days to minimize the risk of causing seizures. Patients requiring abrupt discontinuation should be transitioned to an alternative AEM under close medical supervision. Human patients on rufinamide have experienced status epilepticus in clinical trials at rates of discontinuation of 0.9–4.1%; therefore, caution is warranted. The use of rufinamide is not recommended in patients with severe hepatic impairment. People with mild-tomoderate hepatic impairment may require close monitoring and dosage adjustments, although administration to patients with severe renal impairment is acceptable (Wisniewski, 2010).
Efficacy
Numerous animal studies have shown efficacy against seizures (Bialer et al., 1999). The medication showed strong inhibition of maximal electroshocks, pentylenetetrazol, bicuculline and picrotoxin induced seizures in experimental animals. It delayed kindling and suppressed after discharges in amygdala kindled cats, reduced seizure frequency in rhesus monkeys with chronic alumina foci in motor cortex, and inhibited after discharges in non-kindled cat hippocampus and cortex (Martinez et al., 2012).
Pålhagen and colleagues evaluated the efficacy and safety of adjunctive rufinamide in humans with epilepsy in a 4-week, multi-centre, randomized, placebo-controlled trial comparing escalating doses of rufinamide with placebo (Pålhagen et al., 2001). Patients were randomized to receive either rufinamide, starting at 400 mg/day and increased by 400 mg weekly to a dose of 1600 mg/day (n = 25), or placebo (n = 25). The primary efficacy outcome was seizure frequency ratio during treatment compared to a 28-day baseline period. Baseline frequency was determined retrospectively by participant questionnaire. Patients in the rufinamide group had a median seizure ratio of 0.59 (41%) compared with 1.52 (52%) in the placebo group, a significant reduction after controlling for seizure-free patients. Secondary outcomes of 25% and 50% seizure frequency reduction from baseline showed a significant reduction favouring rufinamide in the former outcome (52% versus 16%) but not the latter (39% versus 16%). In another secondary outcome, patients were identified as having low (£4/28 days) or
Lacosamide, Brivaracetam and Rufinamide
high (>4/28 days) seizure rates, the results of which showed that significantly more patients in the rufinamide group experienced low seizure rates (61% versus 21% during the treatment phase).
The use of rufinamide for adjunctive therapy in human patients with inadequately controlled generalized tonic-clonic seizures was evaluated in a multicentre, randomized, double-blind, placebo-controlled trial (Wisniewski, 2010). Patients ³4 years of age with primary generalized tonic-clonic seizures were randomized to receive either rufinamide 400 mg twice daily or placebo for 140 days. Efficacy was evaluated by the primary outcome of percentage change in seizure frequency per 28 days from baseline. Results did not show a significant difference in the primary outcome. Concomitant AEMs that participants received during the study were not reported. Safety assessments did not show rufinamide to cause more adverse events than placebo. The authors concluded that rufinamide reduced seizure frequency when compared to placebo, but not at a statistically significant rate, and that rufinamide was safe in these patients (Wisniewski, 2010).
A multicentre, randomized, double- blind, placebo-controlled trial was conducted to study the efficacy and safety of rufinamide as an adjunctive agent for the treatment of refractory focal seizures in adolescents and adults (Brodie et al., 2009). After an 8-week baseline phase, patients were randomized to receive either rufinamide (n = 156) or placebo (n = 157) for 13 weeks. Patients on rufinamide started on a dose of 400 mg twice daily, which was titrated to 1600 mg twice daily over 7 days. The primary outcome of this study, the median percentage change in focal seizure frequency during treatment compared to baseline, showed a significant reduction in seizure frequency favouring rufinamide (20.4% median reduction versus 1.6% median increase). Total seizure frequency during the double-blind phase was not significantly reduced by rufinamide, although a post hoc analysis controlling for non-normality of the data demonstrated a significant difference. Another secondary outcome showing significance favouring rufinamide was the percentage of patients achieving a 50% reduction in seizure frequency from baseline (28.2% versus 18.6%). Six rufinamide subjects and three placebo subjects achieved seizure freedom. The authors concluded that rufinamide is an effective and tolerable option for adjunctive treatment in patients experiencing focal seizures refractory to AEMs. The extrapolation of these results is limited to patients with refractory focal seizures on concomitant AEMs, but they demonstrate that rufinamide could be useful in these patients (Wisniewski, 2010).
Summary Recommendations
• Rufinamide has a few specific attributes
that make it an attractive option for an antiseizure medication. Namely, it has few serious side effects, few drug-todrug interactions and is able to be titrated quickly.
• Rufinamide can be given to dogs 2–3 times
daily as an oral medication, but no clinical studies have as yet been performed.
• No information exists on the use of this
drug in cats.
References
Ben-Menachem, E., Biton, V., Jatuzis, D., Abou-Khalil, B., Doty, P. and Rudd, G.D. (2007) Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia 48, 1308–1317.
Beyreuther, B.K., Freitag, J., Heers, C., Krebsfanger, N., Scharfenecker, U. and Stohr, T. (2007) Lacosamide: a review of preclinical properties. CNS Drug Reviews 13, 21–42.
Bialer, M., Johannessen, S.I., Kupferberg, H.J., Levy, R.H., Loiseau, P. and Perucca, E. (1999) Progress report on new antiepileptic drugs: a summary of the fourth Eilat conference (EILAT IV). Epilepsy Research 34, 1–41.
Biton, V., Rosenfeld, W.E., Whitesides, J., Fountain, N.B., Vaiciene, N. and Rudd, G.D. (2008) Intravenous lacosamide as replacement for oral lacosamide in patients with partial-onset seizures. Epilepsia 49, 418–424.
Brandt, C., Heile, A., Potschka, H., Stoehr, T. and Loscher, W. (2006) Effects of the novel antiepileptic drug lacosamide on the development of amygdala kindling in rats. Epilepsia 47, 1803–1809.
Brodie, M.J., Rosenfeld, W.E., Vazquez, B., Sachdeo, R., Perdomo, C., Mann, A. and Arroyo, S. (2009) Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo-controlled trial. Epilepsia 50, 1899–1909.
Carunchio, I., Pieri, M., Ciotti, M.T., Albo, F. and Zona, C. (2007) Modulation of AMPA receptors in cultured cortical neurons induced by the antiepileptic drug levetiracetam. Epilepsia 48, 654–662.
Cawello, W., Boekens, H. and Bonn, R. (2012a) Absorption, disposition, metabolic fate and elimination of the anti-epileptic drug lacosamide in humans: mass balance following intravenous and oral administration. European Journal of Drug Metabolism and Pharmacokinetics 37, 241–248.
Cawello, W., Bonn, R. and Boekens, H. (2012b) Bioequivalence of intravenous and oral formulations of the antiepileptic drug lacosamide. Pharmacology 90, 40–46.
Cawello, W., Bokens, H., Nickel, B., Andreas, J.O. and Halabi, A. (2013) Tolerability, pharmacokinetics, and bioequivalence of the tablet and syrup formulations of lacosamide in plasma, saliva, and urine: saliva as a surrogate of pharmacokinetics in the central compartment. Epilepsia 54, 81–88.
Cheng-Hakimian, A., Anderson, G.D. and Miller, J.W. (2006) Rufinamide: Pharmacology, clinical trials, and role in clinical practice. International Journal of Clinical Practice 60, 1497–1501.
Chinnasami, S., Rathore, C. and Duncan, J.S. (2013) Sinus node dysfunction: An adverse effect of lacosamide. Epilepsia.
Chung, S., Sperling, M.R., Biton, V., Krauss, G., Hebert, D., Rudd, G.D. and Doty, P. (2010) Lacosamide as adjunctive therapy for partial-onset seizures: a randomized controlled trial. Epilepsia 51, 958–967.
Curia, G., Biagini, G., Perucca, E. and Avoli, M. (2009) Lacosamide: a new approach to target voltage-gated sodium currents in epileptic disorders. CNS Drugs 23, 555–568.
Czech, T., Yang, J.W., Csaszar, E., Kappler, J., Baumgartner, C. and Lubec, G. (2004) Reduction of hippocampal collapsin response mediated protein-2 in patients with mesial temporal lobe epilepsy. Neurochem Research 29, 2189–2196.
Errington, A.C., Coyne, L., Stohr, T., Selve, N. and Lees, G. (2006) Seeking a mechanism of action for the novel anticonvulsant lacosamide. Neuropharmacology 50, 1016–1029.
Errington, A.C., Stohr, T., Heers, C. and Lees, G. (2008) The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Molecular Pharmacology 73, 157–169.
Fountain, N.B., Krauss, G., Isojarvi, J., Dilley, D., Doty, P. and Rudd, G.D. (2013) Safety and tolerability of adjunctive lacosamide intravenous loading dose in lacosamide-naive patients with partial-onset seizures. Epilepsia 54, 58–65.
French, J.A., Costantini, C., Brodsky, A. and Von Rosenstiel, P. (2010) Adjunctive brivaracetam for refractory partial-onset seizures: a randomized, controlled trial. Neurology 75, 519–525.
Glauser, T., Kluger, G., Sachdeo, R., Krauss, G., Perdomo, C. and Arroyo, S. (2008) Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology 70, 1950–1958.
Halford, J.J. and Lapointe, M. (2009) Clinical perspectives on lacosamide. Epilepsy Currents 9, 1–9.
Hofler, J. and Trinka, E. (2013) Lacosamide as a new treatment option in status epilepticus. Epilepsia.
Hofler, J., Unterberger, I., Dobesberger, J., Kuchukhidze, G., Walser, G. and Trinka, E. (2011) Intravenous lacosamide in status epilepticus and seizure clusters. Epilepsia 52, e148–152.
Kellinghaus, C., Berning, S., Immisch, I., Larch, J., Rosenow, F., Rossetti, A.O., Tilz, C. and Trinka, E. (2011) Intravenous lacosamide for treatment of status epilepticus. Acta Neurologica Scandinavica 123, 137–141.
Krasowski, M.D. (2010) Therapeutic drug monitoring of the newer anti-epilepsy medications. Pharmaceuticals (Basel) 3, 1909–1935.
Krauss, G., Ben-Menachem, E., Mameniskiene, R., Vaiciene-Magistris, N., Brock, M., Whitesides, J.G. and Johnson, M.E. (2010) Intravenous lacosamide as short-term replacement for oral lacosamide in partial-onset seizures. Epilepsia 51, 951–957.
Krauss, G.L., Edwards, H.B. and Lin, B. (2012) Lacosamide for the treatment of epilepsy. Annals of Medicine 44, 674–679.
Lees, G., Stohr, T. and Errington, A.C. (2006) Stereoselective effects of the novel anticonvulsant lacosamide against 4-AP induced epileptiform activity in rat visual cortex in vitro. Neuropharmacology 50, 98–110.
Lynch, B.A., Lambeng, N., Nocka, K., Kensel-Hammes, P., Bajjalieh, S.M., Matagne, A. and Fuks, B. (2004) The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proceedings of the National Academy of Sciences USA 101, 9861–9866.
Malykh, A.G. and Sadaie, M.R. (2010) Piracetam and piracetam-like drugs: from basic science to novel clinical applications to CNS disorders. Drugs 70, 287–312.
Lacosamide, Brivaracetam and Rufinamide
Martinez, S.E., Bowen, K.A., Remsberg, C.M., Takemoto, J.K., Wright, H.M., Chen-Allen, A.V. and Davies, N.M. (2012) High-performance liquid chromatographic analysis of lacosamide in canine serum using ultraviolet detection: application to pre-clinical pharmacokinetics in dogs. Biomedical Chromatography 26, 606–609.
Matagne, A., Margineanu, D.G., Kenda, B., Michel, P. and Klitgaard, H. (2008) Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. British Journal of Pharmacology 154, 1662–1671.
Pålhagen, S., Canger, R., Henriksen, O., Van Parys, J.A., Riviere, M.E. and Karolchyk, M.A. (2001) Rufinamide: a double-blind, placebo-controlled proof of principle trial in patients with epilepsy. Epilepsy Research 43, 115–124.
Pisani, A., Bonsi, P., Martella, G., De Persis, C., Costa, C., Pisani, F., Bernardi, G. and Calabresi, P. (2004) Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia 45, 719–728.
Rolan, P., Sargentini-Maier, M.L., Pigeolet, E. and Stockis, A. (2008) The pharmacokinetics, CNS pharmacodynamics and adverse event profile of brivaracetam after multiple increasing oral doses in healthy men. British Journal of Clinical Pharmacology 66, 71–75.
Shaibani, A., Biton, V., Rauck, R., Koch, B. and Simpson, J. (2009) Long-term oral lacosamide in painful diabetic neuropathy: a two-year open-label extension trial. European Journal of Pain 13, 458–463.
Stohr, T., Kupferberg, H.J., Stables, J.P., Choi, D., Harris, R.H., Kohn, H., Walton, N. and White, H.S. (2007) Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Research 74, 147–154.
Tai, K.K. and Truong, D.D. (2007) Brivaracetam is superior to levetiracetam in a rat model of post-hypoxic myoclonus. Journal of Neural Transmission 114, 1547–1551.
Van Paesschen, W., Hirsch, E., Johnson, M., Falter, U. and Von Rosenstiel, P. (2013) Efficacy and tolerability of adjunctive brivaracetam in adults with uncontrolled partial-onset seizures: a phase IIb, randomized, controlled trial. Epilepsia 54, 89–97.
Wasterlain, C.G., Stohr, T. and Matagne, A. (2011) The acute and chronic effects of the novel anticonvulsant lacosamide in an experimental model of status epilepticus. Epilepsy Research 94, 10–17.
Willmore, L.J. (2000) Clinical pharmacology of new antiepileptic drugs. Neurology 55, S17–24.
Wisniewski, C.S. (2010) Rufinamide: a new antiepileptic medication for the treatment of seizures associated with lennox-gastaut syndrome. Annals of Pharmacotherapy 44, 658–667.
Wright, H.M., Chen, A.V., Martinez, S.E. and Davies, N.M. (2012) Pharmacokinetics of oral rufinamide in dogs. Journal of Veterinary Pharmacology and Therapeutics 35(6), 529–533.
Yorns, W.R., Jr, Khurana, D.S., Carvalho, K.S., Hardison, H.H., Legido, A. and Valencia, I. (2012) Efficacy of lacosamide as adjunctive therapy in children with refractory epilepsy. Journal of Child Neurology Dec [eprint]
Zona, C., Pieri, M., Carunchio, I., Curcio, L., Klitgaard, H. and Margineanu, D.G. (2010) Brivaracetam (ucb 34714) inhibits Na(+) current in rat cortical neurons in culture. Epilepsy Research 88, 46–54.
21 Benzodiazepines
Simon Platt BVM&S MRCVS Dipl. ACVIM (Neurology) Dipl. ECVN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
Benzodiazepines (BZDs; diazepam, lorazepam, midazolam, clonazepam and clorazepate) are potent, fastacting antiepileptic medications (AEMs) and therefore (particularly diazepam) are the preferred initial therapy in status epilepticus (SE) in animals with no suspected hepatic dysfunction (van der Kleijn et al., 1983; Cascino, 1996; Boothe, 1998; Lowenstein and Alldredge, 1998; Musulin et al., 2011). They are a widely used class of sedative/tranquilizer and anxiolytic agents that differ widely in their time course and their central effects (van der Kleijn et al., 1983).
The basic chemical structure of BZDs is formed from the fusion of a benzene ring and a sevenmembered diazepine ring (Fig. 21.1). The common chemical structure of the BZDs accounts for their similar mechanisms of action. Their differences may be related to pharmacokinetics (van der Kleijn et al., 1983), especially their distribution into and out of the central nervous system, which has been related to the drugs’ lipophilicity and plasma protein binding (van der Kleijn et al., 1983). The lipophilicity of these compounds determines their rapid brain penetration after intravenous administration (van der Kleijn et al., 1983). Although the penetration is rapid, distribution equilibrium among all regions takes longer (van der Kleijn et al., 1983).
General Mechanism of Action
All BZDs share similar neuropharmacologic properties including anxiety reduction, sedation, sleep induction, anticonvulsant effects and muscle relaxation. There are, however, differences among BZDs in affinity for receptor subtypes, which may produce different pharmacologic effects. Thus, some BZDs are more effective than others as anticonvulsants; few of the approximately 35 BZDs available worldwide are used for managing epilepsy. Their primary pharmacologic actions are probably related to a benzodiazepinereceptormediated enhancement of gaminobutyric acid (GABA)ergic transmission, both preand postsynaptically (Treiman, 1989; Nordt and Clark, 1997; Boothe, 1998; Lowenstein and Alldredge, 1998; Singhi et al., 1998). GABA receptors are membranebound proteins that can be divided into two major subtypes: GABAA and GABAB receptors. The ionotropic GABAA receptors are comprised of five subunits forming an integral chloride channel. These receptors are responsible for most inhibitory neurotransmission in the central nervous system (CNS). In contrast, the metabotropic GABAB are G proteincoupled receptors (Fig. 21.2).
Benzodiazepines do not seem to alter the synthesis, release or metabolism of GABA
Benzodiazepines
CH3
O
N
N
Cl
but rather, potentiate its action at the receptor (Riss et al., 2008). The resultant augmented flow of chloride ions into cells decreases the ability of the cell to initiate an action potential (Nordt and Clark, 1997). At higher concentrations, benzodiazepines also limit sustained repetitive neuronal firing, and this effect may be relevant to their mechanism of action in seizures (Lowenstein and Alldredge, 1998; Riss et al., 2008). It seems that benzodiazepines prevent the spread of seizure activity rather than suppress the focus (Treiman, 1989). In animal screening tests, benzodiazepines have a broad spectrum of anticonvulsant activity (Treiman, 1989). They inhibit seizure activity induced by pentylenetetrazol and picrotoxin, being effective at low doses (Treiman, 1989). Other prominent CNS effects of this group of drugs include sedation, hypnosis, decreased anxiety, and muscle relaxation. The benzodiazepines do not produce the same degrees of neuronal depression as barbiturates and volatile anaesthetics. However, as the dose of a benzodiazepine increases, sedation progresses to hypnosis and then to stupor. The existence of multiple benzodiazepine receptors may explain in part the diversity of pharmacological responses.
General Metabolism and Pharmacokinetics
The BZDs also have widely varying pharmacokinetic profiles, with differences in absorption, onset and duration of action and formation of active metabolites. Thus, pharmacokinetic differences often dictate the use of specific BZDs, route(s) of administration and formulation(s).
The inhibitory neurotransmitter GABA binds to the receptor to open the chloride ion gates and produce an inhibitory current. Binding of BZDs to the gsubunit of the receptor is important in the potentiation of GABAergic inhibition. Differentiation between BZDs and GABA is important. Benzodiazepines do not substitute for GABA, but instead enhance the inhibitory effects of GABA. Benzodiazepines allosterically bind to the receptor at a different location than GABA does and enhance the chloride channels’ conductance by increasing the frequency of gated channel opening (Riss et al., 2008). In the search for BZD site ligands with higher therapeutic selectivity and a more favourable safety profile, GABAA receptor subtypes have long been considered promising targets.
In pharmacologic terms, BZD potency refers to the in vivo affinity of the drug (or its active metabolites) for its receptor. Benzodiazepines are classified as low, medium (e.g. clorazepate and diazepam) or high (e.g. clonazepam and lorazepam) potency. Each BZD has a unique pharmacokinetic profile that must be considered when the optimal agent is selected for a particular patient and condition. Key factors to consider include route of administration, rate and extent of absorption, metabolism, formation of active metabolites, elimination and drug interactions.
Most of the benzodiazepines are absorbed completely, with the exception of clorazepate and midazolam. Clorazepate is decarboxylated rapidly in gastric juice before absorption. Midazolam has low oral bioavailability due to metabolism by cytochrome P450 (CYP) enzyme 3A5 in intestinal epithelial tissue, which can reduce the fraction of the dose reaching the bloodstream by up to 50% (Riss et al., 2008). The sum bioavailability (percentage) of the parent BZD diazepam and its metabolites ranges from 74 to 100% in dogs (median 86%) (Loscher and Frey, 1981).
After intravenous administration, BZD pharmacokinetics can be characterized by a multicompartmental mathematical model, with the first phase being distribution, followed by a longer elimination phase. Benzodiazepines also have large volumes of distribution, are highly bound to plasma proteins and readily cross into the placenta. Benzodiazepines
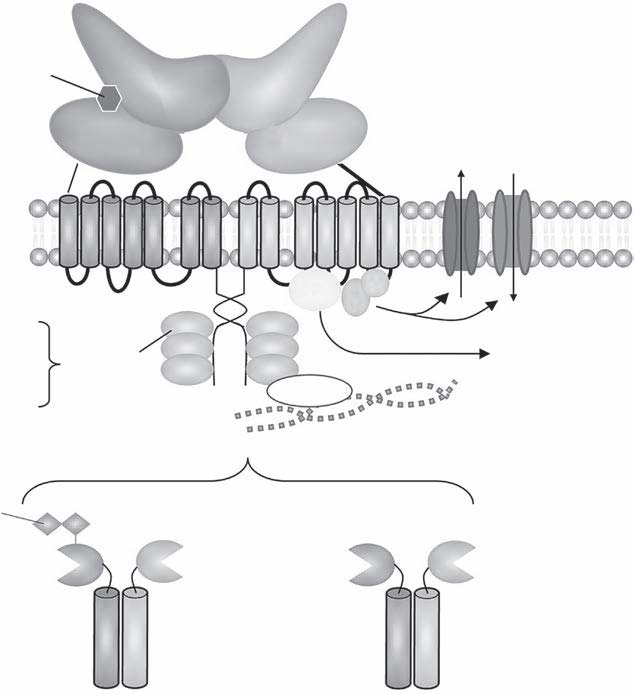

Adhesion molecules Trafficking proteins Transcription factors
Sushi domains
GABAB1a GABAB2 GABAB1b GABAB2
Fig. 21.2a. GABAA receptor.
GABA site
Agonists Antagonists
Barbiturate site
Depressants (also ethanol?) Excitants?
Benzodiazepine site
Agonists (depressants) Antagonists Inverse agonists
Steroid site
Anaesthetics Excitants?
Picrotoxin site
Convulsants Depressants?
Fig. 21.2b. GABAB receptor.
Benzodiazepines
differ in their rates of elimination and the formation of pharmacologically active metabolites. Benzodiazepine metabolism is primarily catalysed by CYPdependent hydroxylation, demethylation and nitroreduction. The CYP isoenzymes catalysing these reactions include 3A4, 3A5, 2B6, 2C9 and 2C19 (Riss et al., 2008). Uridine diphosphate glucuronosyltransferase is also involved in the conjugation of some BZDs. Several BZDs have active metabolites. Diazepam and clorazepate are metabolized into the longacting metabolite Ndesmethyldiazepam (DMD). With multiple doses, the pharmacologic and toxic effects of diazepam are attributable to the parent drug, DMD, and other minor active metabolites
(i.e. temazepam and oxazepam). By contrast, clorazepate undergoes rapid and complete chemical conversion to DMD in the gastrointestinal tract; its pharmacologic effects are largely due to DMD (Riss et al., 2008). NDesmethyldiazepam undergoes glucuronidation to form a glucuronide conjugate (25%) and is hydroxylated (50%) by CYP2C19 and CYP3A4 to form oxazepam. Approximately 5–9% of DMD is excreted unchanged in the urine (Riss et al., 2008).
Benzodiazepines and their metabolites cross the bloodbrain barrier rapidly, although the diffusion rate into the brain varies by drug and is largely determined by lipophilicity. The faster the diffusion rate, the earlier is the onset of pharmacodynamic effects. Peak concentrations usually occur within 15 min of intravenous administration. Rapid entry of BZDs into the CNS and highly perfused tissues is consistent with their short distribution halflives. Following rapid uptake, BZDs redistribute into less wellperfused tissues; the rate of redistribution is fastest for the most lipidsoluble drugs. CSF to plasma drug ratios generally are less than one, although the concentrations in the CSF may stay higher compared to plasma prolonging the duration of effect.
The differences in BZD pharmacokinetics and pharmacodynamics must be considered in order to use these drugs safely and effectively. Equivalent doses of BZDs differ as much as 20fold because of differences in potency. The intensity of singledose effects may vary, even if equipotent doses are used, because of varying oral absorption rates (Ashton, 1994). Duration of action should be considered when choosing a BZD. When maintenance therapy is required (e.g. epilepsy and anxiety), longacting BZDs are preferred because of their prolonged halflife, as effective drug concentrations can be maintained without the need for frequent dosing (Shader et al., 1984).
Pharmacokinetic Interactions and General Adverse Effects
Benzodiazepines interact with other drugs such as certain antidepressants, AEMs (e.g. phenobarbital), sedative antihistamines and opioids, which may result in additive sedative effects (Shader et al., 1984). As discussed earlier, BZD metabolism is complex and largely catalysed by CYP isoenzymes. Consequently, there is potential for interactions between BZDs and drugs that induce or inhibit CYP isoenzymes. The clinical importance of these interactions depends on the net effect of inhibition or induction on the metabolic pathway of a particular BZD. For example, inhibition of a minor pathway may have little impact on drug concentration, whereas inhibition of a major pathway may result in enhanced clinical effect or toxicity. By contrast, addition of an enzymeinducing drug that affects even a relatively minor pathway may lead to a clinically important reduction in plasma BZD concentration. For BZDs with active metabolites, the addition of an inhibitor or inducer may affect only the parent drug, only the metabolite, or both. Clinicians should exercise particular caution when using BZDs with selective serotonin reuptake inhibitors, cimetidine, macrolide antibiotics and antimycotics; these drugs may inhibit reactions catalysed by certain CYP isoenzymes and, therefore, inhibit the metabolism of many BZDs, which results in increased plasma BZD concentrations (Tanaka, 1999). Conversely, potent enzyme inducers (e.g. phenobarbital) substantially increase clearance and reduce the halflife of certain BZDs (Tanaka, 1999).
Although BZDs as a class are well tolerated, veterinarians should be aware of potential adverse effects associated with their use in both dogs and cats. Drowsiness and ‘confusion’ may be indicative of oversedation, a doserelated extension of the sedative/hypnotic effects of BZDs. Paradoxical excitement/hyperactivity is sometimes associated with BZD use and may be more common in cats than dogs. In cats, diazepam specifically has been associated with hepatotoxicity, causing vomiting, depression, jaundice and acute death (Center et al., 1996). Clinical laboratory abnormalities include bilirubin, serum alanine transaminase, aspartate transferase and alkaline phosphatase increases. The toxicity is suggested to be idiosyncratic as it is not experimentally reproducible and is not associated with dose and duration of administration (Center et al., 1996).
In human medicine, dependence refers to the compulsion to take a drug to produce a desired effect or to prevent unpleasant effects that occur when the drug is withheld. Dependence develops in almost onethird of human patients who are treated with BZDs for 4 weeks (Riss et al., 2008). Physical dependence has been described in dogs related to administration of diazepam and lorazepam (McNicholas et al., 1983; Loscher et al., 1989; Sloan et al., 1991a, b, 1993). A withdrawal syndrome upon BZD discontinuation is a common manifestation of BZD dependence in humans and can occur in dogs and cats, necessitating a slow withdrawal if possible (Gatzonis et al., 2000). In dogs, withdrawal signs include biphasic tonic clonic convulsions 24 h later. Withdrawal was seen in dogs after oral administration of increasing doses of diazepam were administered every 8 h for 5–6 weeks. Abstinence was precipitated by administration of a benzodiazepine antagonist, flumazenil, at 0.66, 2, 6, 18, 36 and 72 mg/kg, but included a placebo. Withdrawal intensity increased proportionately with the dose of diazepam and the dose of flumazenil. Seizures only occurred in dogs receiving 9 and 36 mg/kg/day of diazepam. Withdrawal signs increased linearly with plasma and brain concentrations of diazepam, oxezepam and nordiazepam but not with free concentrations of diazepam alone (Sloan et al., 1993).
Substantial differences in adverse event occurrences associated with the BZDs profiled here have not been reported in large randomized, controlled trials comparing BZDs in clinical veterinary patients. Diazepam may have greater effects on respiratory depression than are observed with other BZDs such as lorazepam and midazolam (Treiman, 1989; Cascino, 1996). However, it has been suggested that there is a low overall incidence of respiratory depression with benzodiazepines because of the low density of binding sites in the brainstem (Nordt and Clark, 1997). The dose that causes respiratory arrest in an ill patient may be difficult to determine (Cascino, 1996; Singhi et al., 1998).
Tolerance
Tolerance to antiepileptic medications is associated with a progressive increase in the number and severity of seizures and an increased risk of withdrawal seizures in the presence of a constant maintenance dose. Tolerance to several BZDs has been demonstrated in experimental animal epilepsy models (Frey et al., 1984; Loscher and Schwark, 1985; Scherkl et al., 1988). It is in part because of the development of tolerance that BZDs are generally considered unsuitable for longterm control of epilepsy (Brodie, 1990; Ashton, 1994). Increasing the BZD dose may overcome anticonvulsant tolerance, however, tolerance may recur at the higher dose, and adverse effects may persist or worsen. Crosstolerance between BZDs occurs and appears to be drug specific. RamseyWilliams et al. (1994) demonstrated that after 3 weeks of diazepam treatment, rats developed crosstolerance to the anticonvulsant effects of clobazam, clonazepam and midazolam. Rats treated with midazolam for 3 weeks developed crosstolerance to diazepam but not to clobazam or clonazepam. The authors suggested that differences in tolerance and crosstolerance could result from differential regulation of receptor subunit expression by each drug and from differences between the drugs in their interactions with receptors at the time of
Benzodiazepines
testing. The time to onset of tolerance varies, and the potential of a BZD to induce anticonvulsant tolerance does not appear to have any relationship with its chemical or pharmacokinetic properties (Garratt et al., 1988). Differences in the development of anticonvulsant tolerance have been reported for various BZDs in an amygdalakindling rat model. Young et al. (1988) found that tolerance to clobazam developed within 3 days of initial drug exposure, whereas tolerance to clonazepam developed gradually over the course of a 19day study. Rosenberg et al. (1989) compared the anticonvulsant activity of clonazepam, clobazam and diazepam in rats and found that tolerance developed most rapidly to clobazam and most slowly to clonazepam. In studies investigating anticonvulsant tolerance, animals treated with clorazepate exhibited a later onset or a lesser degree of tolerance than did those treated with either diazepam or clonazepam (Scherkl et al., 1989; Amano et al., 2001). In a study comparing clorazepate and clobazam, tolerance was defined as initial seizure freedom, followed by an increase in seizure frequency to a level greater than that seen with initial treatment.
Tolerance to some side effects of BZDs can also develop. The onset of tolerance to the sedative effects of BZDs usually occurs within 1–2 weeks (Smith and Kroboth, 1987; Kroboth et al., 1990). The differences in time to onset of tolerance to the various pharmacologic effects of BZDs suggest that different mechanisms may be involved (Bateson, 2002). One putative mechanism for the development of tolerance is simple downregulation of GABA receptors in response to prolonged BZD exposure; however, studies testing this hypothesis have reported mixed results (Bateson, 2002). Li et al. (2000) suggested that downregulation of BZD receptor binding sites could not fully explain the development of tolerance because tolerance to some BZDs has been observed even when no changes in BZD receptor binding were noted. Several other mechanisms for BZD tolerance have been proposed on the basis of animal and cell culture models. These processes include uncoupling of allosteric linkage between GABA and BZD sites, changes in the turnover of receptor subunits and changes in receptor gene expression (Bateson, 2002). Considering the evidence that tolerance to the various behavioural effects of BZDs develops at different rates and that behavioural effects of BZDs are mediated by different GABAA receptor subtypes, it was proposed that multiple mechanisms mediate tolerance and dependence (Bateson, 2002). He described a unified model of molecular mechanisms underlying tolerance that incorporates the molecular processes discussed above. This model assumes that initial potentiation of the GABA response leads to desensitization and that prolonged desensitization could result in uncoupling (as either a signal for or a consequence of receptor internalization, i.e. endocytosis). Subsequent to receptor internalization, degradation of certain receptor subunits could provide a signal for changes in GABAA receptor gene transcription. Depending on the receptor subtypes involved, as well as the brain region and neuronal cell types, this model could account for the temporal differences in the development of tolerance to the different effects of BZDs. The reason for differences in time to tolerance between the various BZDs is not entirely clear. The slow onset of tolerance to the anticonvulsant effect of clorazepate could be due to a low degree of intrinsic activity of DMD at the BZD receptor. Gobbi et al. (1987) demonstrated that diazepam and DMD have the same affinity for the central type of BZD receptors, but when the intrinsic activity of DMD was calculated, it proved to be a partial agonist, with intrinsic activity approximately 43% that of diazepam.
Diazepam
Diazepam is a Schedule IV drug under the 1970 Controlled Substances Act, but is not approved for use in animals by the FDA (Boothe, 1998). However, it remains the first drug of choice for the treatment of SE in dogs and cats, unless there is evident hepatic dysfunction (Boothe, 1998). Diazepam is usually administered intravenously but can also be given intramuscularly; however, its absorption is unpredictable when administered this way.
Metabolism and pharmacokinetics
The drug is well absorbed after oral administration but undergoes rapid and extensive hepatic metabolism. Although only 1–3% of diazepam is orally bioavailable (compared to 7–21% after IV administration), 74% to 100% of the drug and all active metabolites are available (Frey and Loscher, 1985). The generation of active metabolites complicates the utility of therapeutic monitoring for benzodiazepines as a guide to therapy because anticonvulsant activity is not necessarily correlated with the concentration of the parent compound. Rectal and nasal administration of diazepam has also been described (Loscher and Frey, 1981; Papich and Alcorn, 1995; Platt et al., 2000; Musulin et al., 2011).
The major metabolites of diazepam, nordiazapam (desmethyldiazepam) and oxazepam, have up to 33% of the activity of the parent drug (Boothe, 1998). 3Hydroxydiazepam and temazepam are also metabolites, although their anticonvulsant activity is not known (Frey and Loscher, 1985; Nordt and Clark, 1997). Diazepam is metabolized to nordiazepam by CYP2B11 and nordiazepam is metabolized to oxazepam by CYP3A12 in dogs, while diazepam is metabolized to temazepam by CYP3A12 and temazepam is metabolized to oxazepam by CYP2B11 in dogs (Shou et al., 2003; Lu et al., 2005).
The terminal halflife of diazepam following IV administration is approximately
0.25 h in the dog and the halflives of the individual metabolites range from 2.5 to 5.2 h (Klotz et al., 1976; Loscher and Frey, 1981; Papich and Alcorn, 1995; Musulin et al., 2011). The reported plasma clearance of diazepam in nongreyhound dogs ranges from 11.5 to 60 ml/min/kg (Papich and Alcorn, 1995; Musulin et al., 2011). The reported range of diazepam volume of distribution in nongreyhound dogs is 1.4–3.5 l/kg. The mean volume of distribution seen after an IV bolus
(0.5 mg/kg) of diazepam in greyhounds was determined to be 2.448 l/kg with a range of 1.974–3.083 l/kg (KuKanich and Nauss, 2012). The mean Cl was 27.9 ml/min/kg with a range of 22.4–44.9 ml/min/kg. The mean T½ was 1.0 h with a range of 0.5–1.4 h. With respect to the diazepam metabolites, temazepam was not detected above the lower limit of analytical quantification, 5 ng/ml (KuKanich and Nauss, 2012). In addition, the mean Cmax of oxazepam was 44.7 ng/ml with a range of 31.7–58.1 ng/ml at a mean Tmax of 0.79 h. The mean Cmax of nordiazepam was 305.2 ng/ml with a range of 249.0–342.0 ng/ml at a mean Tmax of 0.36 h (KuKanich and Nauss, 2012).
The high concentrations of nordiazepam seen in greyhounds following IV diazepam are suggestive that greyhounds are able to efficiently metabolize diazepam by a mechanism previously identified as CYP2B11 in dogs (KuKanich and Nauss, 2012). However, the specific metabolizing enzymes in greyhounds have not been identified, and the rapid elimination of diazepam and formation of nordiazepam may occur by CYP2B11 or a different enzyme.
Rectal administration of 2 mg/kg diazepam in dogs yielded peak diazepam concentrations of only 75 ng/ml; this compared to peak desmethyldiazepam and oxazepam concentrations of approximately 1600 and 550 ng/ml, respectively (Papich and Alcorn, 1995). Both metabolites’ concentrations remained above 300 ng/ml at the 8h study end (Papich and Alcorn, 1995). As such, rectal administration of diazepam can be expected to yield effective antiepileptic activity, with concentrations remaining above the minimum recommended for about 8 h for desmethyldiazepam (Papich and Alcorn, 1995). In clinically affected seizuring dogs untreated with other medications, a rectal diazepam dose of 1 mg/kg resulted in a mean time to peak plasma concentration of
14.3 min and the peak benzodiazepine concentration reached in the plasma was 474 mg/l (Podell, 1996). However, the bioavailability of rectal diazepam has been documented to be only 2.7% for low (0.5 mg/kg) and 7.4% for high (2.0 mg/kg) doses of diazepam, using HPLC for analysis (Papich and Alcorn, 1995). Total benzodiazepine bioavailability levels were 66.0% and 79.9% after low and high doses, respectively. A recent study evaluated the pharmacokinetics of diazepam administered
Benzodiazepines
per rectum via compounded (i.e. not commercially available) suppositories (Probst et al., 2013). The study aimed to determine whether a dose of 2 mg/kg of this formulation would result in plasma concentrations shown to be effective for control of SE or cluster seizures (i.e. 150 to 300 ng/ml) in dogs within a clinically useful interval (10–15 min). Six healthy mixedbreed dogs were randomly assigned to two groups of three dogs each in a crossoverdesign study. Diazepam (2 mg/kg) was administered IV or via suppository per rectum, and blood samples were collected at predetermined time points. Following a 6 or 7day washout period, each group received the alternate treatment. Plasma concentrations of diazepam and nordiazepam were analysed via reversed phase highperformance liquid chromatography. Plasma concentrations of diazepam and nordiazepam exceeded the targeted range £3 min after IV administration in all dogs. After suppository administration, targeted concentrations of diazepam were not detected in any dogs, and targeted concentrations of nordiazepam were detected after 90 min (n = 2 dogs) or 120 min (n = 3) or were not achieved (n = 1). On the basis of these results, administration of 2 mg diazepam/kg via the compounded suppositories was not recommended for emergency treatment of seizures in dogs (Probst et al., 2013).
In dogs, following intranasal administration of 0.5 mg/kg diazepam, bioavailability approximated 40% in one study (Papich and Alcorn, 1995), with the bioavailability of diazepam and all of its metabolites approximated 80 ± 9% in another (Platt et al., 2000). Mean peak plasma concentration of benzodiazepine was 448 ± 41 ng/ml at 4.5 min, compared with 1316 ± 216 at 3 min when given IV (Platt et al., 2000). Maximum concentrations were achieved within 10 min after nasal administration. Additionally, plasma drug concentrations exceeded the recommended anticonvulsant therapeutic concentration of 300 ng/ml when delivered intranasally (Platt et al., 2000). A recent study investigated the pharmacokinetics of diazepam following IV, INdrop and INatomized administration in dogs (Musulin et al., 2011). Six dogs were administered diazepam (0.5 mg/kg) via all three routes following a randomized block design. Plasma samples were collected and concentrations of diazepam and its active metabolites, oxazepam and desmethyldiazepam, were quantified with highperformance liquid chromatography (HPLC). Mean diazepam concentrations >300 ng/ml were reached within 5 min in both IN groups. Diazepam was converted into its metabolites within 5 and 10 min, respectively, after IV and IN administration. The halflives of the metabolites were longer than that of the parent drug after both routes of administration. The bioavailability of diazepam after INdrop and atomizednasal administration was 42% and 41%, respectively (Musulin et al., 2011). These values exceed previously published bioavailability data for rectal administration of diazepam in dogs.
Diazepam and nordiazepam have been studied in cats after intravenous administration of 5, 10 and 20 mg/kg of diazepam and 5 and 10 mg/kg of nordiazepam (Cotler et al., 1984). Elimination of both drugs was linear over the range of doses covered. Total body clearance of diazepam (4.72 ± 2.45 ml/min/kg) was sixfold greater than that of nordiazepam
(0.85 ± 0.25 ml/min/kg). Approximately 50% of an administered dose of diazepam was biotransformed to nordiazepam in the cat (Cotler et al., 1984).
Pharmacokinetic interactions and adverse reactions
General interactions and adverse reactions have been discussed in the first part of this chapter. Specific to diazepam, there is a concern of hepatotoxicity in cats associated with the use of diazepam, which is potentially reversible; in addition to its associated behavioural sideeffects this drug has fallen out of favour as an antiepileptic drug in this species (Center et al., 1996; Park, 2012). Manifestations of hepatotoxicity in cats administered this drug include vomiting, depression, jaundice, lethargy and acute death (Center et al., 1996). This side effect appears to be idiosyncratic causing increased serum alanine transaminase, aspartate transferase and alkaline phosphatase activities (Center et al., 1996).
Dosing and monitoring recommendations
With its relatively brief duration of action and rapid development of tolerance, diazepam is not a definitive therapy for chronic seizures in dogs. In fact, tolerance in this species to the anticonvulsant activity of diazepam develops within a week. However, because IV diazepam produces transiently high serum and brain concentrations of the drug, with its rapid transport across the bloodbrain barrier, it can be a useful drug therapy. Because SE may end spontaneously, IV diazepam should not be administered to a patient presenting in a postictal state unless there is another seizure. For chronic seizure control in cats, it can be given orally in a dose of 2 to 5 mg two or three times daily and increased by 2 mg at a time. Longterm efficacy in this species may be due to high concentrations of nordiazepam being detectable throughout all the brain matter (Placidi et al., 1976).
It has been recommended to use 0.5 to
1.0 mg/kg intravenously, up to a maximum total dose of 20 mg, in dogs and cats (Boothe, 1998). This dose can be repeated to effect or twice within 2 h (Boothe, 1998). Constant rate intravenous infusions of diazepam have been advocated in human and veterinary patients (Boothe, 1998). The recommended dose is 2–5 mg/h in 5% dextrose in water (Boothe, 1998). Continuous diazepam infusions have been shown to be a reasonably effective modality to control refractory SE in children and has been associated with a reduced need for ventilatory and vasopressor support (Singhi et al., 1998). If the diazepam does not control the seizures, the use of phenobarbital should be considered. Probably the most common and most dangerous error made in the management of SE is to treat repeated seizures with repeated doses of IV diazepam without treating the precipitating factors definitively and without administering an adequate loading dose of a longeracting antiepileptic drug. In this situation, the patient will continue to have seizures, toxic concentrations of diazepam or diazepam metabolites will accumulate, and serious morbidity may result from diazepam overdosage. Alternative strategies for emergency antiepileptic therapy when diazepam use is individually inappropriate are detailed in Chapter 24.
Rectal administration of diazepam may be considered initially at a dose of 0.5–
2.0 mg/kg body weight depending upon whether the animal was being treated with phenobarbital before the onset of SE (Podell, 1996; Boothe, 1998). Diazepam metabolism is upregulated in epileptic patients on hepatic enzymeinducing medications such as phenobarbital. Therefore, it may be necessary to use the higher dose in dogs receiving longterm phenobarbital therapy (Wagner et al., 1998). Although rectal administration of diazepam is often recommended for athome therapy in patients with cluster seizures, this route is not efficacious in some patients, and is awkward for pet owners. Recent evaluation of a rectal suppositorycompounded formulation of diazepam has suggested that it does not reach therapeutic concentrations for the purposes of stopping seizure activity in dogs (Probst et al., 2013). Proposed explanations for the variable clinical efficacy of rectal diazepam include unsuccessful delivery because of the presence of rectal faeces, expulsion of drug, unpredictable absorption and extensive firstpass hepatic metabolism.
Intranasal administration is currently an option but can be practically difficult without a commercially available high concentration preparation. The dose recommended would be the same as that for rectal administration and has been shown to rapidly reach presumed therapeutic antiseizure serum concentrations.
Efficacy
Diazepam is a drug of first choice for treatment of early SE and cluster seizures (acute repetitive seizures). It can be administered as an IV bolus, as a continuous infusion, nasally or rectally, which enhances its utility in managing seizure emergencies. Randomized controlled human trials support diazepam as a drug of first choice for managing SE. No such data exist in veterinary medicine but it is an established first line treatment for SE in most practices.
Benzodiazepines
Success rates of IV diazepam for treating SE in humans vary. In a randomized doubleblind study comparing diazepam and lorazepam, Leppik et al. (1983) found that 76% of SE episodes (25 of 33) were terminated by one or two diazepam doses (5 mg/min). A total of 384 patients with overt SE and 134 patients with subtle SE were randomly assigned to receive either diazepam (0.15 mg/kg) followed by phenytoin (18 mg/kg), lorazepam (0.1 mg/kg) alone, phenobarbital (15 mg/kg) alone or phenytoin (18 mg/kg) alone. Treatment with diazepam plus phenytoin was successful in 55.8% of people (53 of 95) with overt SE and 8.3% of people (3 of 36) with subtle SE (Treiman et al., 1998). Alldredge et al. (2001) conducted a randomized double blind trial to determine the effectiveness of IV diazepam, lorazepam and placebo on SE in people when the drugs were administered by paramedics before patients arrived at the hospital. They found that SE was terminated by the time of arrival in the emergency department in 42.6% of the 68 patients treated with one or two 5mg doses of IV diazepam (infused over 1–2 min). Again, no such studies exist in veterinary medicine at this time.
The use of diazepam per rectum (RDZ) in the home to control generalized cluster seizures in 11 dogs diagnosed with idiopathic epilepsy was evaluated by Podell (1995) over a 16month period. All dogs had a prior history of clusters of generalized seizures and were treated with multiple antiepileptic drugs. Owners were instructed to administer diazepam injectable solution (5 mg/ml) per rectum to their dogs at a dose of 0.5 mg/kg when an initial generalized seizure occurred and when a second or third generalized seizure occurred within 24 h of the first seizure. Seizure activity was recorded by owners in a daily log before the onset of RDZ use and for the duration of RDZ use, which ranged from 57 to 464 days (median = 157 days). The median age at which the first seizure occurred and the median age at the time of enrolment in the study were 19 and 42 months, respectively. All 11 dogs were treated with phenobarbital, with ten dogs receiving concomitant bromide therapy. No significant correlation between the duration of the first, second, or third antiepileptic drug therapy and the change in the number of cluster seizure events before or after use of RDZ was found. Comparisons of seizure activity were done for the same time interval before and after the onset of RDZ availability. A significant decrease in the total number of seizure events and the total number of cluster seizure events was found after RDZ availability. Similarly, a significant difference in the average number of seizures per cluster seizure event and the total number of isolated seizure events occurred before and after RDZ therapy (Podell, 1995).
Continuous IV infusions of diazepam can be safe and effective based on the human and experimental literature (DelgadoEscueta and EnrileBacsal, 1983). Use of IV diazepam can result in seizure relapse within 2 h of a single injection in approximately 50% of human patients. Therefore, multiple injections or continuous infusion may be required, which can lead to drug accumulation and possibly to acute respiratory depression, sedation and hypotension. The development of tolerance has also been reported for infusions lasting >24 h.
Midazolam
Midazolam is a more recently developed watersoluble benzodiazepine of the group of 1,2annelated benzodiazepines (Koul et al., 1997). The water solubility allows midazolam to be packaged without diluents such as propylene glycol, thus decreasing venous irritation and making it a more suitable drug to administer intranasally than diazepam (Nordt and Clark, 1997). At physiologic pH, midazolam becomes extremely lipophilic, enabling a rapid onset of action (Nordt and Clark, 1997). Midazolam solubility is achieved when the injectable solution is buffered to a pH of 2.9–3.7 (Scott et al., 1999; Lahat et al., 2000a; Riss et al., 2008). A humanapproved midazolam injectable product (Versed; Roche Laboratories, Nutley, New Jersey) is available, but there are at present no approved veterinary midazolam preparations. A dose for cats and dogs, which is poorly documented, is 0.07–0.2 mg/kg IM or IV.
Metabolism and Pharmacokinetics
Like other benzodiazepines, midazolam is biotransformed by hepatic microsomal oxidation followed by glucuronide conjugation (Nordt and Clark, 1997). Alphahydroxymidazolam, the primary metabolite after hydroxylation, is pharmacologically active and has sedative properties equivalent to midazolam in humans, although only low levels of this metabolite could be detected after IV or IM administration of midazolam to dogs (Court and Greenblatt, 1992; Nordt and Clark, 1997). All the metabolites are rapidly excreted in the urine in humans, but in dogs a more predominant extrarenal excretion is suggested, probably through the bile (Court and Greenblatt, 1992). In dogs the elimination halflife of these metabolites is 11 min (Court and Greenblatt, 1992). Midazolam has been shown to have a wide margin of safety and a broad therapeutic index (Walsh and DelgadoEscueta, 1993). It will diffuse rapidly across the capillary wall into the CNS and can be mixed with saline or glucose solutions (Koul et al., 1997). The mean plasma elimination halflife in dogs was shown to be 53–77 min following IV administration (Court and Greenblatt, 1992).
Unlike diazepam, with erratic and incomplete intramuscular absorption, midazolam is rapidly absorbed following IM injection, with a high bioavailability, an early onset of sedation and early clinical effects (Nordt and Clark, 1997). The peak plasma concentration in dogs after IM administration was seen within 15 min, and a mean elimination halflife of 56 min was demonstrated with a bioavailability of more than 90% (Court and Greenblatt, 1992). These properties may support the use of IM midazolam for dogs with seizures. A more recent study has also supported this view (Schwartz et al., 2013). This study compared the pharmacokinetics of midazolam after IV, IM and rectal (PR) administration. Six healthy dogs were administered 0.2 mg/ kg midazolam IV, IM or PR in a randomized, threeway crossover design with a 3day washout between study periods. Blood samples were collected at baseline and at predetermined intervals until 480 min after administration. Plasma midazolam concentrations were measured by highpressure liquid chromatography with UV detection. Rectal administration resulted in erratic systemic availability with undetectable to low plasma concentrations. Arithmetic mean values ± SD for midazolam peak plasma concentrations were 0.86 ± 0.36 mg/ml (C0) and 0.20 ± 0.06 mg/ ml (Cmax) following IV and IM administration, respectively. Time to peak concentration (Tmax) after IM administration was 7.8 ± 2.4 min with a bioavailability of 50 ± 16% (Schwartz et al., 2013).
Rectal administration of the parenteral midazolam solution achieved concentrations that would be considered therapeutic in humans in a recent study by the author (Eagleson et al., 2012); however, the Tmax was prolonged (39 ±
14.49 min). The bioavailability determined was 49%, which is improved from a previous report (Court and Greenblatt, 1992) revealing almost no absorption! Pharmacokinetic reports of rectal midazolam in humans are variable with bioavailability ranging from 18% to 52% and a Tmax of 12.1 – 31 min (Clausen et al., 1988; Payne et al., 1989; Malinovsky et al., 1993, 1995). Rectal diazepam in dogs has been shown to have a bioavailability from 51.7 (± 21.8%) to 79.9 (± 20.7%) and a Tmax of 14.3 min (± 3.7) (Mealey and Boothe, 1995; Papich and Alcorn, 1995; Eagleson et al., 2012). This may well be based on whether the midazolam passes into the caudal and middle rectal veins avoiding firstpass metabolism or whether administered further cranially passing into the cranial rectal veins which drain into the liver by way of the portal vein (Schwartz et al., 2013). Based on these pharmacokinetics, rectal diazepam would seem to be superior to rectal midazolam as an anticonvulsant.
Pharmacokinetic studies in humans evaluating intranasal administration of the parenteral 5 mg/ml formulation at doses of
0.2 mg/kg have demonstrated mean peak plasma concentrations between 104 ng/ml and 182 ng/ml within 12 min, well above the 40 ng/ml considered to be the minimum therapeutic level for sedation in adults (Allonen et al., 1981; Rey et al., 1991; Malinovsky et al., 1993). Estimated bioavailability of the parenteral 5 mg/ml formulation administered at
0.2 mg/kg intranasally in humans ranges from 50 to 83% (Rey et al., 1991; Burstein et al., 1997; Knoester et al., 2002). Intranasal midazolam
Benzodiazepines
at 0.2 mg/kg has been demonstrated to penetrate the human brain in 2–5 min, as shown by the appearance of beta activity in the EEG and suppressed epileptic activity (O’Regan et al., 1996). This suggests that early entry of modest midazolam concentrations into the brain compartment has antiseizure effect and that the plasma midazolam concentration required to stop a seizure could be far less than the concentration needed to produce sedation (Wermeling et al., 2009). Intranasal administration of midazolam in one canine study yielded maximum plasma concentration within 15 min, but the dose given was not reported (Lui et al., 1991). Another pharmacokinetic study in dogs of intranasal midazolam administered at 1.5 mg/kg found a bioavailability of only 10% (Henry et al., 1998). The author has conducted an evaluation of a novel midazolam gel (4% hydroxypropyl methylcellulose) and demonstrated it to be readily absorbed when administered intranasally with a pharmacokinetic profile superior than the parenteral solution given IN or PR to dogs (Eagleson et al., 2012). The mean peak plasma concentration of 450 ng/ml seen was more than twice the peak plasma concentrations seen in human studies using the parenteral solution at the same dose via IN route. The T and C of the intranasal gel were
maxmax
comparable to IM administration of a 0.5 mg/ kg dose in dogs (C, 549 ± 121 ng/ml; T,
maxmax
8 ± 2 min) (Court and Greenblatt, 1992). Alternative intranasal formulations of midazolam have demonstrated favourable pharmacokinetics in humans. The common feature to all the experimental formulations is a solubility enhancer, resulting in a more concentrated product allowing delivery of a much smaller volume. The aim is to generate a product that could deliver an effective dose in 0.1–0.2 ml, an accepted retention volume in the human nose (Romeo et al., 1998). Due to the extreme phenotypical variation seen in the domestic dog (in both size of animal and conformation of the head), an average nasal retention volume in dogs is difficult to define. None the less, a highly concentrated/low volume product would be advantageous in brachycephalic and largebreed dogs. The recent study evaluated the highest concentration (50 mg/ml) to date, with 30 mg/ml being the closest highest concentration previously reported (Haschke et al., 2010; Eagleson et al., 2012). Comparisons between gel formulations studied in humans are difficult. All pharmacokinetic studies in humans evaluating alternative midazolam formulations have used doses less than 0.1 mg/kg. The highest peak plasma concentration reported in these studies is 80 ng/ml reached within 7.2 or 10.3 min (Wermeling et al., 2006; Haschke et al., 2010). Reported bioavailabilities of the alternative formulations have ranged anywhere from 64 to 92% (Gudmundsdottir et al., 2001; Haschke et al., 2010).
Buccal absorption of midazolam has been demonstrated in dogs; however, the dose was not reported and the conditions under which the experiment was conducted were not clinically applicable to dogs with SE (Zhang et al., 2002).
Pharmacokinetic interactions and adverse reactions
General interactions and adverse reactions have been discussed in the first part of this chapter.
Dosing and monitoring recommendations
The typical dose administered to dogs and cats is 0.07 to 0.2 mg/kg intramuscular, intranasal or intravenous. Based on some of the aforementioned pharmacokinetic studies, a slightly higher dose may be warranted to achieve an anticonvulsive effect.
Efficacy
Midazolam has a significant antiepileptic effect in humans proven to be caused by GABAA receptor stimulation (Cascino, 1996; Scott et al., 1998), and has been shown to be more effective and safer for the control of seizures than comparable doses of diazepam (Koul et al., 1997; Nordt and Clark, 1997).
In the treatment of SE, midazolam can be administered by IV bolus, continuous IV infusion or IM injection. Clinical experience with midazolam for treating SE (as initial treatment or for refractory SE) is limited. A recent human study demonstrated the beneficial effects of a midazolam intravenous infusion in 11 of 12 patients with refractory SE (Koul et al., 1997).
In three controlled human clinical trials, the efficacy of intranasal midazolam was similar to or better than that of IV or rectal diazepam (Lahat et al., 2000b; Fisgin et al., 2002; Mahmoudian and Zadeh, 2004). Midazolam has also been found to be safe and effective when administered as a continuous infusion to treat refractory generalized convulsive SE (Kumar and Bleck, 1992; Parent and Lowenstein, 1994; Koul et al., 2002; Singhi et al., 2002). In a randomized human trial comparing buccal midazolam with rectal diazepam in children, the drugs showed similar efficacy and onset of action (Scott et al., 1999). In that study, seizure cessation was achieved in 75% of cases (30 of 40) with midazolam and in 59% of cases (23 of 39) with diazepam treatment (P = 0.16). Results from another randomized controlled human trial that compared buccal midazolam with rectal diazepam for emergency treatment of seizures in children suggested that midazolam was more effective than diazepam (McIntyre et al., 2005). Therapeutic success (defined as cessation of visible signs of seizure activity within 10 min of drug administration, lack of respiratory depression and no further seizures within 1 h) was noted in 56% of midazolamtreated patients (61 of 109) and 27% of diazepamtreated patients (30 of 110), a 29% difference between groups (95% confidence interval, 16–41%). Openlabel human studies have suggested that intranasal midazolam is safe and effective for acute seizure management in children (Jeannet et al., 1999; Fisgin et al., 2000, 2002; Lahat et al., 2000a; Mahmoudian and Zadeh, 2004). In a prospective, randomized, openlabel human study, intranasal midazolam was as safe and effective as was IV diazepam for managing seizures, with 88% of seizures (23 of 26) responding to initial treatment with midazolam and 92% (24 of 26) responding to diazepam (Lahat et al., 2000a).
Midazolam has great potential as a shortacting anticonvulsant in dogs, possessing twice the affinity for the benzodiazepine receptor and four times the hypnotic potency of diazepam (Reves et al., 1985). However, the precise midazolam plasma concentration needed to stop seizure activity is not known in dogs or humans, although the anticonvulsant effect of midazolam has been documented in experimental animal models (Koul et al., 1997; Nordt and Clark, 1997). Knowledge of midazolam’s effectiveness as an anticonvulsant in dogs is limited to a single study, which demonstrated 0.2 mg/kg of IV midazolam had a stronger suppressive effect against lidocaineinduced seizures than the same dose of diazepam (Horikawa et al., 1990).
Lorazepam
Metabolism and pharmacokinetics
Lorazepam is a longacting benzodiazepine that interacts with a high degree of affinity for the GABA receptor complex. This highaffinity binding in turn leads to a prolonged duration of action in humans. Lorazepam is not as lipidsoluble as diazepam, and in animals, brain and cerebrospinal fluid concentrations of lorazepam rise at a slower rate than those of diazepam after IV injection (Lowenstein and Alldredge, 1998).
A veterinary study evaluated the pharmacokinetic properties of lorazepam administered IV and rectally in normal dogs (Podell et al., 1998). Three adult dogs received
0.2 mg/kg IV lorazepam. The rectal administration phase was conducted after a 10day drugfree period, at which time one dog each received either 0.5, 0.75 or 1 mg/kg rectal lorazepam through a 3.8 cm plastic administrator attached to a syringe. Blood samples were collected at 0, 5, 15 (20 for IV), 30, 45, 60, 90, 120, 240 and 360 min after administration in heparinized glass tubes and kept on ice until the plasma was harvested and stored at −80°C. Plasma concentrations of lorazepam were determined by high performance liquid chromatography. Peak plasma lorazepam concentrations were reached within 5 min in all dogs after IV administration. Concentrations remained above 190 ng/ml for 20 min in two dogs, and above 30 ng/ml for 90 min in all dogs.
Benzodiazepines
As lorazepam concentrations in the brain remain elevated longer than in plasma, the therapeutic effect may persist longer than these times. Subjectively, all dogs became sedate within 5 min of the injection for a duration of ≈90 min. Administration of lorazepam rectally yielded plasma concentrations less than the lower limit of detection (5 ng/ml) in all dogs at all times, regardless of dose administered. A hydrolysis procedure using bglucuronidase was performed on these samples to determine if the drug was metabolized to its inactive metabolite, lorazepam glucuronide. Remeasurement of lorazepam concentrations resulted in higher plasma concentrations at all times for each dose indicating that an extensive first pass effect must have occurred.
Pharmacokinetic interactions and adverse reactions
General interactions and adverse reactions have been discussed the first part of this chapter.
Dosing and monitoring recommendations
In humans, lorazepam is generally given as an IV bolus at doses of 0.05–0.1 mg/kg over 2 min, and the dose may be repeated in 10 min. Lorazepam can reach presumed therapeutic blood concentrations in dogs when administered IV at 0.2 mg/kg (Podell et al., 1998). Pilot studies using 0.5–1.0 mg/kg rectal lorazepam in normal dogs however, suggested that lorazepam may not be acceptable for rectal administration because of an extensive first pass effect (Podell et al., 1998).
Efficacy
Large lorazepam doses have been used as an alternative to pentobarbital for treating refractory SE; for all nine human cases in an openlabel study, lorazepam terminated SE (Labar et al., 1994). Results from four comparative studies (three of blinded design) have suggested that lorazepam is superior to phenytoin and as effective as clonazepam, diazepam or the combination of diazepam and phenytoin in the initial treatment of SE (Sorel et al., 1981; Leppik et al., 1983; Treiman et al., 1998; Alldredge et al., 2001). In the previously mentioned human study by Alldredge, IV lorazepam (2 mg, one to two doses) administered to 66 adults with repetitive or ongoing generalized seizures lasting >5 min terminated convulsions by the time of arrival at the emergency department in 59.1% of patients (Alldredge et al., 2001). Similarly, Leppik et al. (1983) found that one or two lorazepam doses (4 mg each) terminated seizures in 89% of SE episodes.
Clinical studies in children have been mostly unblinded and have included retrospective and prospective designs. In a prospective openlabel study, Appleton et al. (1995) compared IV or rectal lorazepam and diazepam treatments in 86 children. A single dose terminated seizures in 76% of patients treated with lorazepam and in 51% of patients treated with diazepam; the difference between treatments was not statistically significant. Qureshi et al. (2002) performed a comparative audit of IV lorazepam and diazepam. The authors suggested that lorazepam is probably as effective as diazepam is for stopping acute seizures in children. Seizures were controlled within 15 min in 65% of diazepamtreated patients (11 of 17) (median time 3 min) and in 65% of lorazepamtreated patients (20 of 31) (median time 5 min).
One veterinary study compared the duration of seizure control of lorazepam with diazepam in 16 dogs presenting in SE or with active cluster seizures (Naeser et al., 2004). Previous seizure history and anticonvulsant therapy was not a consideration for inclusion into this study. Animals were excluded if there was a known metabolic, toxic or traumatic cause of the seizure. Dogs were randomly assigned to receive either lorazepam
(0.2 mg/kg IV) or diazepam (0.5 mg/kg IV), and the clinicians were blinded as to which drug they were administering. The duration of the study was 12 h from the time of drug administration, and the animals were monitored for any indication of seizure activity, including generalized motor activity, focal motor activity (e.g. movement of facial or limb musculature) and change in the level of consciousness. The study ended at 12 h poststudy drug administration or when the dog seizured before the end of the 12h study period. The results indicated no significant difference between lorazepam versus diazepam with regard to median seizurefree interval (2.8 h for diazepam versus 3.4 h for lorazepam, p = 0.58 by log rank test), or with regard to percentage seizurefree for the duration of the observation period (1/8 for lorazepam versus 3/8 for diazepam, p = 0.51 by Fisher’s exact test). There was also no difference between the two drugs regarding the number of animals in which seizures were initially controlled (6/8 in each group). Lorazepam used at this dose does not appear to result in a significant increase in duration of seizure control for dogs with SE and cluster seizures.
Clorazepate
Clorazepate (clorazepate dipotassium) is a benzodiazepine prodrug that acts by enhancing GABA activity in the brain. This drug is now not available in the UK but it can be found in the USA and other countries as 3.75 and
7.5 mg extendedrelease tablets and standardrelease 3.75, 7.5 and 15 mg tablets.
Metabolism and pharmacokinetics
Nordiazepam is the active metabolite produced following rapid hydrolysation, which occurs in the stomach when the drug is administered orally. The serum elimination halflife of nordiazepam after clorazepate administration is 46 h in dogs but may be as short as 3 h. After oral administration of 2 mg/kg, clorazepate reaches peak benzodiazepine concentration of 446 to 1542 ng/ml in dogs; mean residence time is 8.5 h but this increases with multiple administrations to approximately 12 h (Forrester et al., 1990). Oral doses as high as 2 mg/kg can result in profound sedation and ataxia but such signs may resolve 3 to 4 days after treatment (Scherkl et al., 1989). Although the drug is available as a regular and sustained release formulation, there seems to be no difference in the pharmacokinetics in dogs between the two (Brown and Forrester, 1991).
Pharmacokinetic interactions and adverse reactions
General interactions and adverse reactions for BZD have been discussed in the first part of this chapter. Anecdotally, clorazepate is an alternative antiepileptic drug in cats but because it is an active metabolite of diazepam in other species, its role in diazepaminduced hepatotoxicity cannot be ruled out. Therefore the author does not recommend the use of this drug in cats unless absolutely necessary. Clorazepate should not be combined with phenobarbital as there is anecdotal evidence of a risk of hepatotoxicity and drug interactions causing an unpredictable antiepileptic effect.
Tolerance to the use of this drug has been described in some dogs with resultant reduction in its anticonvulsant properties (Scherkl et al., 1989). Withdrawal seizures have also been described following the use and cessation of this drug.
Dosing and monitoring recommendations
Currently the author uses this drug as a temporary antiepileptic therapy for cluster seizures (0.5–2.0 mg/kg PO tid; see Chapter 23) or during the load period of another maintenance antiepileptic medication such as bromide.
Efficacy
The effectiveness of adjunctive clorazepate for various treatmentresistant seizures has been demonstrated in humans; however, the clinical experience and data available are less extensive for clorazepate than for other BZDs (Trimble, 2002). An openlabel human clinical study of clorazepate therapy suggested that the drug might be useful as an addon treatment for generalized major and minor seizures
(e.g. absence and myoclonic) (Booker, 1974). In that study, an excellent response (defined
Benzodiazepines
as complete seizure control) was observed for 19% of patients with major generalized seizures (three of 16), 39% of patients with absence seizures (seven of 18), 33% of patients with akinetic seizures (three of nine) and 57% of patients with myoclonic seizures (four of seven). Other doubleblind and openlabel studies have demonstrated the efficacy of adjunctive clorazepate for both focal and generalized seizures (Berchou et al., 1981; Wilensky et al., 1981; Fujii et al., 1987). There have also been good results with clorazepate in the treatment of refractory childhood epilepsies. In an openlabel study by Naidu et al., 11 children with generalized seizures (absence and atonic) who received clorazepate as monotherapy (n = 4) or as adjunctive therapy with valproate (n = 7) had a reduction in the number of clinically observed seizures (Naidu et al., 1986; Riss et al., 2008). In another openlabel study, clorazepate was added to standard AEDs in 29 children with various seizure disorders; 72% of patients experienced improved seizure control (Riss et al., 2008). There are no published trials evaluating the efficacy of clorazepate in veterinary patients.
Summary Recommendations
• Benzodiazepines are short-acting anti-epileptic medications when used intravenously as a bolus or by constantrate infusion for the treatment of SE and cluster seizures.
• Benzodiazepines are ineffective as anti-epileptic medications when given orally in the dog, except for clorazepate, and only on a shortterm basis.
• Benzodiazepines can also be given rectally, buccally and intranasally for emergency seizure control. Rectal administration does not always avoid firstpass metabolism through the liver and so is erratic in its effect.
• Clorazepate can be given orally q8h to dogs for the shortterm control of seizure activity, but tolerance will develop.
References
Alldredge, B.K., Gelb, A.M., Isaacs, S.M., Corry, M.D., Allen, F., Ulrich, S., Gottwald, M.D., O’Neil, N., Neuhaus, J.M., Segal, M.R. and Lowenstein, D.H. (2001) A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. New England Journal of Medicine 345, 631–637.
Allonen, H., Ziegler, G. and Klotz, U. (1981) Midazolam kinetics. Clinical Pharmacology and Therapeutics 30, 653–661.
Amano, K., Katsuragi, S., Takamatsu, J., Ogata, A., Miyazaki, C., Deshimaru, M. and Miyakawa, T. (2001) Differences between the tolerance characteristics of two anticonvulsant benzodiazepines in the amygdaloid-kindled rat. Life Sciences 69, 1049–1055.
Appleton, R., Sweeney, A., Choonara, I., Robson, J. and Molyneux, E. (1995) Lorazepam versus diazepam in the acute treatment of epileptic seizures and status epilepticus. Developmental Medicine and Child Neurology 37, 682–688.
Ashton, H. (1994) Guidelines for the rational use of benzodiazepines. When and what to use. Drugs 48, 25–40.
Bateson, A.N. (2002) Basic pharmacologic mechanisms involved in benzodiazepine tolerance and withdrawal. Current Pharmaceutical Design 8, 5–21.
Berchou, R.C., Rodin, E.A. and Russell, M.E. (1981) Clorazepate therapy for refractory seizures. Neurology 31, 1483–1485.
Booker, H.E. (1974) Clorazepate dipotassium in the treatment of intractable epilepsy. Journal of the American Medical Association 229, 552–555.
Boothe, D.M. (1998) Anticonvulsant therapy in small animals. Veterinary Clinics of North America - Small Animal Practice 28, 411–448.
Brodie, M.J. (1990) Established anticonvulsants and treatment of refractory epilepsy. Lancet 336, 350–354.
Brown, S.A. and Forrester, S.D. (1991) Serum disposition of oral clorazepate from regular-release and sustained-delivery tablets in dogs. Journal of Veterinary Pharmacology and Therapeutics 14, 426–429.
Burstein, A.H., Modica, R., Hatton, M., Forrest, A. and Gengo, F.M. (1997) Pharmacokinetics and pharmacodynamics of midazolam after intranasal administration. Journal of Clinical Pharmacology 37, 711–718.
Cascino, G.D. (1996) Generalized convulsive status epilepticus. Mayo Clinic Proceedings 71, 787–792.
Center, S.A., Elston, T.H., Rowland, P.H., Rosen, D.K., Reitz, B.L., Brunt, J.E., Rodan, I., House, J., Bank, S., Lynch, L.R., Dring, L.A. and Levy, J.K. (1996) Fulminant hepatic failure associated with oral administration of diazepam in 11 cats. Journal of the American Veterinary Medical Association 209, 618–625.
Clausen, T.G., Wolff, J., Hansen, P.B., Larsen, F., Rasmussen, S.N., Dixon, J.S. and Crevoisier, C. (1988) Pharmacokinetics of midazolam and alpha-hydroxy-midazolam following rectal and intravenous administration. British Journal of Clinical Pharmacology 25, 457–463.
Cotler, S., Gustafson, J.H. and Colburn, W.A. (1984) Pharmacokinetics of diazepam and nordiazepam in the cat. Journal of Pharmacological Science 73, 348–351.
Court, M.H. and Greenblatt, D.J. (1992) Pharmacokinetics and preliminary observations of behavioral changes following administration of midazolam to dogs. Journal of Veterinary Pharmacology and Therapeutics 15, 343–350.
Delgado-Escueta, A.V. and Enrile-Bacsal, F. (1983) Combination therapy for status epilepticus: intravenous diazepam and phenytoin. Advances in Neurology 34, 477–485.
Eagleson, J.S., Platt, S.R., Strong, D.L., Kent, M., Freeman, A.C., Nghiem, P.P., Zheng, B. and White, C.A. (2012) Bioavailability of a novel midazolam gel after intranasal administration in dogs. American Journal of Veterinary Research 73, 539–545.
Fisgin, T., Gurer, Y., Senbil, N., Tezic, T., Zorlu, P., Okuyaz, C. and Akgun, D. (2000) Nasal midazolam effects on childhood acute seizures. Journal of Child Neurology 15, 833–835.
Fisgin, T., Gurer, Y., Tezic, T., Senbil, N., Zorlu, P., Okuyaz, C. and Akgun, D. (2002) Effects of intranasal midazolam and rectal diazepam on acute convulsions in children: prospective randomized study. Journal of Child Neurology 17, 123–126.
Forrester, S.D., Brown, S.A., Lees, G.E. and Hartsfield, S.M. (1990) Disposition of clorazepate in dogs after single- and multiple-dose oral administration. American Journal of Veterinary Research 51, 2001–2005.
Frey, H.H. and Loscher, W. (1985) Pharmacokinetics of anti-epileptic drugs in the dog: a review. Journal of Veterinary Pharmacology and Therapeutics 8, 219–233.
Frey, H.H., Philippin, H.P. and Scheuler, W. (1984) Development of tolerance to the anticonvulsant effect of diazepam in dogs. European Journal of Pharmacology 104, 27–38.
Fujii, T., Okuno, T., Go, T., Ochi, J., Hattori, H., Kataoka, K. and Mikawa, H. (1987) Clorazepate therapy for intractable epilepsy. Brain and Development 9, 288–291.
Garratt, J.C., Gent, J.P., Feely, M. and Haigh, J.R. (1988) Can benzodiazepines be classified by characterising their anticonvulsant tolerance-inducing potential? European Journal of Pharmacology 145, 75–80.
Gatzonis, S.D., Angelopoulos, E.K., Daskalopoulou, E.G., Mantouvalos, V., Chioni, A., Zournas, C. and Siafakas, A. (2000) Convulsive status epilepticus following abrupt high-dose benzodiazepine discontinuation. Drug Alcohol Depend 59, 95–97.
Gobbi, M., Barone, D., Mennini, T. and Garattini, S. (1987) Diazepam and desmethyldiazepam differ in their affinities and efficacies at ‘central’ and ‘peripheral’ benzodiazepine receptors. Journal of Pharmacy and Pharmacology 39, 388–391.
Gudmundsdottir, H., Sigurjonsdottir, J.F., Masson, M., Fjalldal, O., Stefansson, E. and Loftsson, T. (2001) Intranasal administration of midazolam in a cyclodextrin based formulation: bioavailability and clinical evaluation in humans. Pharmazie 56, 963–966.
Haschke, M., Suter, K., Hofmann, S., Witschi, R., Frohlich, J., Imanidis, G., Drewe, J., Briellmann, T.A., Dussy, F.E., Krahenbuhl, S. and Surber, C. (2010) Pharmacokinetics and pharmacodynamics of nasally delivered midazolam. British Journal of Clinical Pharmacology 69, 607–616.
Henry, R.J., Ruano, N., Casto, D. and Wolf, R.H. (1998) A pharmacokinetic study of midazolam in dogs: nasal drop vs. atomizer administration. Pediatric Dentistry 20, 321–326.
Horikawa, H., Tada, T., Sakai, M., Karube, T. and Ichiyanagi, K. (1990) Effects of midazolam on the threshold of lidocaine-induced seizures in the dog – comparison with diazepam. Journal of Anesthesia 4, 265–269.
Jeannet, P.Y., Roulet, E., Maeder-Ingvar, M., Gehri, M., Jutzi, A. and Deonna, T. (1999) Home and hospital treatment of acute seizures in children with nasal midazolam. European Journal of Paediatric Neurology 3, 73–77.
Klotz, U., Antonin, K.H. and Bieck, P.R. (1976) Pharmacokinetics and plasma binding of diazepam in man, dog, rabbit, guinea pig and rat. Journal of Pharmacology and Experimental Therapeutics 199, 67–73.
Knoester, P.D., Jonker, D.M., Van Der Hoeven, R.T., Vermeij, T.A., Edelbroek, P.M., Brekelmans, G.J. and De Haan, G.J. (2002) Pharmacokinetics and pharmacodynamics of midazolam administered as a concentrated intranasal spray. A study in healthy volunteers. British Journal of Clinical Pharmacology 53, 501–507.
Benzodiazepines
Koul, R.L., Raj Aithala, G., Chacko, A., Joshi, R. and Seif Elbualy, M. (1997) Continuous midazolam infusion as treatment of status epilepticus. Archives of Disease in Childhood 76, 445–448.
Koul, R., Chacko, A., Javed, H. and Al Riyami, K. (2002) Eight-year study of childhood status epilepticus: midazolam infusion in management and outcome. Journal of Child Neurology 17, 908–910.
Kroboth, P.D., Mcauley, J.W. and Smith, R.B. (1990) Alprazolam in the elderly: pharmacokinetics and pharmacodynamics during multiple dosing. Psychopharmacology (Berlin) 100, 477–484.
KuKanich, B. and Nauss, J.L. (2012) Pharmacokinetics of the cytochrome P-450 substrates phenytoin, theophylline, and diazepam in healthy Greyhound dogs. Journal of Veterinary Pharmacology and Therapeutics 35, 275–281.
Kumar, A. and Bleck, T.P. (1992) Intravenous midazolam for the treatment of refractory status epilepticus. Critical Care Medicine 20, 483–488.
Labar, D.R., Ali, A. and Root, J. (1994) High-dose intravenous lorazepam for the treatment of refractory status epilepticus. Neurology 44, 1400–1403.
Lahat, E., Goldman, M., Barr, J., Bistritzer, T. and Berkovitch, M. (2000a) Comparison of intranasal midazolam with intravenous diazepam for treating febrile seizures in children: prospective randomised study. British Medical Journal 321, 83–86.
Lahat, E., Goldman, M., Barr, J., Bistritzer, T. and Berkovitch, M. (2000b) Intranasal midazolam as a treatment of autonomic crisis in patients with familial dysautonomia. Pediatric Neurology 22, 19–22.
Leppik, I.E., Derivan, A.T., Homan, R.W., Walker, J., Ramsay, R.E. and Patrick, B. (1983) Double-blind study of lorazepam and diazepam in status epilepticus. Journal of the American Medical Association 249, 1452–1454.
Li, M., Szabo, A. and Rosenberg, H.C. (2000) Down-regulation of benzodiazepine binding to alpha 5 subunit-containing gamma-aminobutyric Acid(A) receptors in tolerant rat brain indicates particular involvement of the hippocampal CA1 region. Journal of Pharmacology and Experimental Therapeutics 295, 689–696.
Loscher, W. and Frey, H.H. (1981) Pharmacokinetics of diazepam in the dog. Archives Internationale de Pharmacodynamie et de Thérapie 254, 180–195.
Loscher, W. and Schwark, W.S. (1985) Development of tolerance to the anticonvulsant effect of diazepam in amygdala-kindled rats. Experimental Neurology 90, 373–384.
Loscher, W., Honack, D. and Fassbender, C.P. (1989) Physical dependence on diazepam in the dog: precipitation of different abstinence syndromes by the benzodiazepine receptor antagonists Ro 15-1788 and ZK 93426. British Journal of Pharmacology 97, 843–852.
Lowenstein, D.H. and Alldredge, B.K. (1998) Status epilepticus. New England Journal of Medicine 338, 970–976.
Lu, P., Singh, S.B., Carr, B.A., Fang, Y., Xiang, C.D., Rushmore, T.H., Rodrigues, A.D. and Shou, M. (2005) Selective inhibition of dog hepatic CYP2B11 and CYP3A12. Journal of Pharmacology and Experimental Therapeutics 313, 518–528.
Lui, C.Y., Amidon, G.L. and Goldberg, A. (1991) Intranasal absorption of flurazepam, midazolam, and triazolam in dogs. Journal of Pharmacological Science 80, 1125–1129.
Mahmoudian, T. and Zadeh, M.M. (2004) Comparison of intranasal midazolam with intravenous diazepam for treating acute seizures in children. Epilepsy and Behavior 5, 253–255.
Malinovsky, J.M., Lejus, C., Servin, F., Lepage, J.Y., Le Normand, Y., Testa, S., Cozian, A. and Pinaud, M. (1993) Plasma concentrations of midazolam after i.v., nasal or rectal administration in children. British Journal of Anaesthesia 70, 617–620.
Malinovsky, J.M., Populaire, C., Cozian, A., Lepage, J.Y., Lejus, C. and Pinaud, M. (1995) Premedication with midazolam in children. Effect of intranasal, rectal and oral routes on plasma midazolam concentrations. Anaesthesia 50, 351–354.
McIntyre, J., Robertson, S., Norris, E., Appleton, R., Whitehouse, W.P., Phillips, B., Martland, T., Berry, K., Collier, J., Smith, S. and Choonara, I. (2005) Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: a randomised controlled trial. Lancet 366, 205–210.
McNicholas, L.F., Martin, W.R. and Cherian, S. (1983) Physical dependence on diazepam and lorazepam in the dog. Journal of Pharmacology and Experimental Therapeutics 226, 783–789.
Mealey, K.L. and Boothe, D.M. (1995) Bioavailability of benzodiazepines following rectal administration of diazepam in dogs. Journal of Veterinary Pharmacology and Therapeutics 18, 72–74.
Musulin, S.E., Mariani, C.L. and Papich, M.G. (2011) Diazepam pharmacokinetics after nasal drop and atomized nasal administration in dogs. Journal of Veterinary Pharmacology and Therapeutics 34, 17–24.
Naeser, J., Lichtenberger, M., Mariani, C., Thamm, D.H.M. and Kirby, R. (2004) Clinical comparison of lorazepam vs. diazepam in the control of canine seizures. Journal of Veterinary Emergency and Critical Care 14, S1–S17.
Naidu, S., Gruener, G. and Brazis, P. (1986) Excellent results with clorazepate in recalcitrant childhood epilepsies. Pediatric Neurology 2, 18–22.
Nordt, S.P. and Clark, R.F. (1997) Midazolam: a review of therapeutic uses and toxicity. Journal of Emergency Medicine 15, 357–365.
O’Regan, M.E., Brown, J.K. and Clarke, M. (1996) Nasal rather than rectal benzodiazepines in the management of acute childhood seizures? Developmental Medicine and Child Neurology 38, 1037–1045.
Papich, M.G. and Alcorn, J. (1995) Absorption of diazepam after its rectal administration in dogs. American Journal of Veterinary Research 56, 1629–1636.
Parent, J.M. and Lowenstein, D.H. (1994) Treatment of refractory generalized status epilepticus with continuous infusion of midazolam. Neurology 44, 1837–1840.
Park, F.M. (2012) Successful treatment of hepatic failure secondary to diazepam administration in a cat. Journal of Feline Medical Surgery 14, 158–160.
Payne, K., Mattheyse, F.J., Liebenberg, D. and Dawes, T. (1989) The pharmacokinetics of midazolam in paediatric patients. European Journal of Clinical Pharmacology 37, 267–272.
Placidi, G.F., Tognoni, G., Pacifici, G.M., Cassano, G.B. and Morselli, P.L. (1976) Regional distribution of diazepam and its metabolites in the brain of cat after chronic treatment. Psychopharmacology (Berlin) 48, 133–137.
Platt, S.R., Randell, S.C., Scott, K.C., Chrisman, C.L., Hill, R.C. and Gronwall, R.R. (2000) Comparison of plasma benzodiazepine concentrations following intranasal and intravenous administration of diazepam to dogs. American Journal of Veterinary Research 61, 651–654.
Podell, M. (1995) The use of diazepam per rectum at home for the acute management of cluster seizures in dogs. Journal of Veterinary Internal Medicine 9, 68–74.
Podell, M. (1996) Seizures in dogs. Veterinary Clinics of North America - Small Animal Practice 26, 779–809.
Podell, M., Wagner, S.O. and Sams, R.A. (1998) Lorazepam concentrations in plasma following its intravenous and rectal administration in dogs. Journal of Veterinary Pharmacology and Therapeutics 21, 158–160.
Probst, C.W., Thomas, W.B., Moyers, T.D., Martin, T. and Cox, S. (2013) Evaluation of plasma diazepam and nordiazepam concentrations following administration of diazepam intravenously or via suppository per rectum in dogs. American Journal of Veterinary Research 74, 611–615.
Qureshi, A., Wassmer, E., Davies, P., Berry, K. and Whitehouse, W.P. (2002) Comparative audit of intravenous lorazepam and diazepam in the emergency treatment of convulsive status epilepticus in children. Seizure 11, 141–144.
Ramsey-Williams, V.A., Wu, Y. and Rosenberg, H.C. (1994) Comparison of anticonvulsant tolerance, crosstolerance, and benzodiazepine receptor binding following chronic treatment with diazepam or midazolam. Pharmacology, Biochemistry, and Behavior 48, 765–772.
Reves, J.G., Fragen, R.J., Vinik, H.R. and Greenblatt, D.J. (1985) Midazolam: pharmacology and uses. Anesthesiology 62, 310–324.
Rey, E., Delaunay, L., Pons, G., Murat, I., Richard, M.O., Saint-Maurice, C. and Olive, G. (1991) Pharmacokinetics of midazolam in children: comparative study of intranasal and intravenous administration. European Journal of Clinical Pharmacology 41, 355–357.
Riss, J., Cloyd, J., Gates, J. and Collins, S. (2008) Benzodiazepines in epilepsy: pharmacology and pharmacokinetics. Acta Neurologica Scandanavica 118, 69–86.
Romeo, V.D., Demeireles, J., Sileno, A.P., Pimplaskar, H.K. and Behl, C.R. (1998) Effects of physicochemical properties and other factors on systemic nasal drug delivery. Advanced Drug Delivery Reviews 29, 89–116.
Rosenberg, H.C., Tietz, E.I. and Chiu, T.H. (1989) Tolerance to anticonvulsant effects of diazepam, clonazepam, and clobazam in amygdala-kindled rats. Epilepsia 30, 276–285.
Scherkl, R., Kurudi, D. and Frey, H.H. (1988) Tolerance to the anticonvulsant effect of clorazepate and clonazepam in mice. Pharmacology and Toxicology 62, 38–41.
Scherkl, R., Kurudi, D. and Frey, H.H. (1989) Clorazepate in dogs: tolerance to the anticonvulsant effect and signs of physical dependence. Epilepsy Research 3, 144–150.
Schwartz, M., Munana, K.R., Nettifee-Osborne, J.A., Messenger, K.M. and Papich, M.G. (2013) The pharmacokinetics of midazolam after intravenous, intramuscular, and rectal administration in healthy dogs. Journal of Veterinary Pharmacology and Therapeutics 36(5), 471–477.
Scott, R.C., Besag, F.M., Boyd, S.G., Berry, D. and Neville, B.G. (1998) Buccal absorption of midazolam: pharmacokinetics and EEG pharmacodynamics. Epilepsia 39, 290–294.
Scott, R.C., Besag, F.M. and Neville, B.G. (1999) Buccal midazolam and rectal diazepam for treatment of prolonged seizures in childhood and adolescence: a randomised trial. Lancet 353, 623–626.
Benzodiazepines
Shader, R.I., Pary, R.J., Harmatz, J.S., Allison, S., Locniskar, A. and Greenblatt, D.J. (1984) Plasma concentrations and clinical effects after single oral doses of prazepam, clorazepate, and diazepam. Journal of Clinical Psychiatry 45, 411–413.
Shou, M., Norcross, R., Sandig, G., Lu, P., Li, Y., Lin, Y., Mei, Q., Rodrigues, A.D. and Rushmore, T.H. (2003) Substrate specificity and kinetic properties of seven heterologously expressed dog cytochromes p450. Drug Metabolism and Disposition 31, 1161–1169.
Singhi, S., Banerjee, S. and Singhi, P. (1998) Refractory status epilepticus in children: role of continuous diazepam infusion. Journal of Child Neurology 13, 23–26.
Singhi, S., Murthy, A., Singhi, P. and Jayashree, M. (2002) Continuous midazolam versus diazepam infusion for refractory convulsive status epilepticus. Journal of Child Neurology 17, 106–110.
Sloan, J.W., Martin, W.R., Wala, E. and Dickey, K.M. (1991a) Chronic administration of and dependence on halazepam, diazepam, and nordiazepam in the dog. Drug and Alcohol Dependence 28, 249–264.
Sloan, J.W., Martin, W.R. and Wala, E.P. (1991b) A comparison of the physical dependence inducing properties of flunitrazepam and diazepam. Pharmacology, Biochemistry, and Behavior 39, 395–405.
Sloan, J.W., Martin, W.R. and Wala, E. (1993) Effect of the chronic dose of diazepam on the intensity and characteristics of the precipitated abstinence syndrome in the dog. Journal of Pharmacology and Experimental Therapeutics 265, 1152–1162.
Smith, R.B. and Kroboth, P.D. (1987) Influence of dosing regimen on alprazolam and metabolite serum concentrations and tolerance to sedative and psychomotor effects. Psychopharmacology (Berlin) 93, 105–112.
Sorel, L., Mechler, L. and Harmant, J. (1981) Comparative trial of intravenous lorazepam and clonazepam im status epilepticus. Clinical Therapeutics 4, 326–336.
Tanaka, E. (1999) Clinically significant pharmacokinetic drug interactions with benzodiazepines. Journal of Clinical Pharmacy and Therapeutics 24, 347–355.
Treiman, D.M. (1989) Pharmacokinetics and clinical use of benzodiazepines in the management of status epilepticus. Epilepsia 30(Suppl. 2), S4–10.
Treiman, D.M., Meyers, P.D., Walton, N.Y., Collins, J.F., Colling, C., Rowan, A.J., Handforth, A., Faught, E., Calabrese, V.P., Uthman, B.M., Ramsay, R.E. and Mamdani, M.B. (1998) A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. New England Journal of Medicine 339, 792–798.
Trimble, M.R. (2002) On the use of tranquillisers in epilepsy. Epilepsia 43(Suppl. 2), 25–27.
Van Der Kleijn, E., Baars, A.M., Vree, T.B. and Van Der Dries, A. (1983) Clinical pharmacokinetics of drugs used in the treatment of status epilepticus. Advances in Neurology 34, 421–440.
Wagner, S.O., Sams, R.A. and Podell, M. (1998) Chronic phenobarbital therapy reduces plasma benzodiazepine concentrations after intravenous and rectal administration of diazepam in the dog. Journal of Veterinary Pharmacology and Therapeutics 21, 335–341.
Walsh, G.O. and Delgado-Escueta, A.V. (1993) Status epilepticus. Neurologic Clinics 11, 835–856.
Wermeling, D.P., Record, K.A., Kelly, T.H., Archer, S.M., Clinch, T. and Rudy, A.C. (2006) Pharmacokinetics and pharmacodynamics of a new intranasal midazolam formulation in healthy volunteers. Anesthesia and Analgesia 103, 344–349, table of contents.
Wermeling, D.P., Record, K.A., Archer, S.M. and Rudy, A.C. (2009) A pharmacokinetic and pharmacodynamic study, in healthy volunteers, of a rapidly absorbed intranasal midazolam formulation. Epilepsy Research 83, 124–132.
Wilensky, A.J., Ojemann, L.M., Temkin, N.R., Troupin, A.S. and Dodrill, C.B. (1981) Clorazepate and phenobarbital as antiepileptic drugs: a double-blind study. Neurology 31, 1271–1276.
Young, N.A., Lewis, S.J., Harris, Q.L., Jarrott, B. and Vajda, F.J. (1988) Differences in the development of tolerance to two anticonvulsant benzodiazepines in the amygdaloid kindled rat. Journal of Pharmacy and Pharmacology 40, 365–367.
Zhang, J., Niu, S., Zhang, H. and Streisand, J.B. (2002) Oral mucosal absorption of midazolam in dogs is strongly pH dependent. Journal of Pharmacological Science 91, 980–982.
22 Imepitoin (Pexion®)
Luisa De Risio
Neurology/Neurosurgery Unit, Centre for Small Animal Studies, Animal Health Trust, Newmarket, UK
Imepitoin (formerly named AWD131-138 or ELB138; 1-(4-chlorophenyl)-4-morpholinoimidazolin-2-one) is a next generation anti-epileptic medication (AEM) (Fig. 22.1). It was originally developed from a series of imidazolinones in the 1990s. Following Phase I clinical studies, further clinical development for use in epileptic people was suspended because of metabolic differences between smokers and nonsmokers (Löscher et al., 2013). However, due to anticonvulsant and anxiolytic activity exhibited in rodent and dog model studies, it was further developed for clinical use in idiopathic epileptic dogs (Bialer et al., 2013; Rundfeldt and Löscher 2013). Imepitoin has been registered as Pexion® by Boehringer Ingelheim and licensed throughout the European Union on 26 February 2013 for use in idiopathic epileptic dogs with generalized seizures. Pexion® is available as 100 mg and 400 mg oval, white, half-scored tablets in bottles of 100 and 250 (SPC, 2013).
Mechanism of Action
Imepitoin is a low affinity partial agonist for the BZD binding site of g-aminobutyric acid (GABA)A receptor (Rostock et al., 1998a; Sigel et al., 1998; Rundfeldt and Löscher 2013). By binding at this site imepitoin potentiates the amplitude of GABA-evoked chloride (Cl−) currents resulting in neuronal hyperpolarization (Fig. 22.2). The GABA-potentiating anxiolytic and anticonvulsant effects of imepitoin could be counteracted by the BZD antagonist flumazenil, indicating that the interaction of imepitoin with the BZD binding site may be imepitoin’s main mechanism of action (Sigel et al., 1998).
In addition imepitoin blocks voltage-activated Ca2+ channels in a dose-dependent manner (Bialer et al., 1999). It is as yet unclear which channel subtype is affected and how this activity may contribute to imepitoin’s anticonvulsant activity. Furthermore, imepitoin antagonizes the increased firing of action potentials induced by corticotrophin-releasing factor in locus coeruleus neurons of murine brainstem slices. This effect could contribute to imepitoin’s anxiolytic activity (Rostock et al., 1998a).
Metabolism and Pharmacokinetics
Pharmacokinetic studies in dogs indicate that imepitoin is well absorbed (>92%) after oral administration and that no pronounced first pass effect occurs. After oral administration of imepitoin tablets at 30 mg/kg without food, peak plasma concentrations are attained
Imepitoin 497
rapidly with a time to maximum concentration (Tmax) of 1.8 ± 0.49 h and a maximum concentration (Cmax) of 15.63 ± 5.21 mg/ml. Co-administration of imepitoin tablets with food reduces the total area under the curve by 30% but produces no significant change in Tand C. Gender-specific differences
max max
do not occur in dogs. Dose linearity occurs when imepitoin is administered at a dosage between 10 and 30 mg/kg BID. Volume of distribution is 1.2 ± 0.63 l/kg. Imepitoin crosses the blood-brain barrier rapidly and in a relatively high concentration without involvement of active transport or active clearance. The in vivo plasma protein binding of imepitoin in dogs is 60–70%. No accumulation of imepitoin in plasma occurs after repeated administration, as steady state is reached
O N N
N O
Cl

after the first dose. Imepitoin is extensively metabolized by oxidation in the liver prior to elimination. Metabolite profiles in urine and faeces revealed four major inactive metabolites. Imepitoin is rapidly cleared from blood (clearance = 8.27 ± 3.07 ml/min/kg) with an elimination half-life of 1.7 ± 0.57 h. The majority of imepitoin and its metabolites are excreted via the faecal route rather than the urinary route so that no major change in pharmacokinetics and no accumulation are expected in renally impaired dogs (Boehringer Ingelheim, data on file). Pharmacokinetic parameters of imepitoin after oral administration in healthy beagle dogs are summarized in Table 22.1.
Pharmacokinetic Interactions and Adverse Reactions
There is no information on pharmacokinetic interactions between imepitoin and other medications. No harmful clinical interactions were observed when imepitoin was used as adjunctive treatment to phenobarbital (PB) or primidone (PRM) in a small number of epileptic dogs (Rieck et al., 2006). Although

Fig. 22.2. Neuronal receptor targets for imepitoin.
Table 22.1. Pharmacokinetic parameters of imepitoin at steady state in healthy beagle dogs administered 30 mg/kg BID orally (Boehringer Ingelheim, data on file 2008).
Dose AUCinf Cl/F Cmax MRTinf administered (h µg/ml) Vz/F (l/kg) (ml/min/kg) (µg/ml) Tmax (h) T½ (h) (h)
30 mg/kg Mean ± 65.39 ± 21.86 1.2 ± 0.63 8.27 ± 3.08 15.63 ± 5.21 1.8 ± 0.49 1.7 ± 0.57 3.3 ± 0.58 PO BID SD (steady Min 26.14 0.42 4.35 7.66 1 0.8 2.2 state) Median 64.85 1.09 7.53 14.2 1.8 1.6 3.2
Max 114.83 2.53 17.15 28.3 3 2.9 5
PO, per os; AUCinf, area under the curve from dosing time extrapolated to infinity; Vz/F, apparent volume of distribution of the area fraction of the dose absorbed; Cl/F, plasma clearance per fraction of the dose absorbed; Cmax, maximum concentration; Tmax, time to maximum concentration; T½, elimination half-life; MRTinf, mean residence time extrapolated to infinity.
imepitoin is a low affinity partial agonist for the benzodiazepine (BZD) binding site of the GABAA receptor it has not prevented the pharmacological activity of full BZD agonists such as diazepam in the clinical setting (e.g. dogs in status epilepticus) (Boehringer Ingelheim, data on file 2011).
The following mild and generally transient adverse reactions have been observed (in order of decreasing frequency) in pre-clinical and clinical studies in dogs administered 10–30 mg/kg BID: polyphagia at the beginning of the treatment, hyperactivity, polyuria, polydipsia, somnolence, hypersalivation, emesis, ataxia, lethargy, diarrhoea, prolapsed nictitating membrane, decreased vision and sensitivity to sound (Löscher et al., 2004; Rieck et al., 2006; Boehringer Ingelheim, data on file; SPC, 2013).
Repeated overdose of up to five times the highest recommended dose (= 150 mg/kg BID) in healthy experimental dogs resulted in central nervous system (e.g. lethargy, abnormal nystagmus) and gastrointestinal-related effects and reversible prolongation of the QT interval. These clinical signs were not usually life-threatening and generally resolved within 24 h if symptomatic treatment was given. In addition, decreased body weight was observed in some dogs. In male dogs administered ten times the upper recommended therapeutic dose (= 300 mg/kg BID), diffuse atrophy of seminiferous tubules in the testes and associated decreased sperm counts were seen (SPC, 2013).
In 116 idiopathic epileptic dogs administered imepitoin at 10–30 mg/kg for up to 20 weeks, serum ALT, AP, AST, GGT, GLDH and BUN remained within reference ranges, however a slight increase in creatinine concentrations compared to baseline was observed. This increase did not result in creatinine concentrations above the reference range and was not associated with any clinically significant observations or events (SPC, 2013).
Tolerance did not develop and no withdrawal signs were observed after treatment discontinuation in epileptic dogs administered an average imepitoin dosage of 20 mg/kg BID for up to 51 months (Löscher et al., 2004). In addition, no withdrawal signs were observed after abrupt termination of imepitoin administered at very high doses (215 mg/kg/day) for 13 weeks during a toxicity study in beagle dogs (Schneider, 2001). Withdrawal signs were only seen when a high dose of the BZD antagonist flumazenil was injected intravenously in experimental dogs after treatment with imepitoin for 5 weeks (Löscher et al., 2004).
Dosing and Monitoring Recommendations
The dosage of imepitoin recommended by the manufacturer is 10 mg/kg to 30 mg/kg orally every 12 h. Although the plasma half- life of imepitoin is approximately 2 h the results seen in seizure models and multiple field studies in dogs demonstrate that the pharmacodynamic effect is longer than the pharmacokinetic half-life. The recommended initial dosage of imepitoin is 10 mg/kg every 12 h. If seizure control is not satisfactory after at least
Imepitoin 499
1 week of treatment at this dose and the medication is well tolerated, the dose can be increased by 50 to 100% increments up to a maximum 30 mg/kg every 12 h. As imepitoin bioavailability is greater when administered to fasted dogs, the manufacturer recommendation is to keep a consistent timing of tablet administration in relation to feeding.
Reference range of plasma or serum imepitoin concentration is unknown and there are no therapeutic monitoring recommendations for imepitoin from the manufacturer. Pharmacokinetic studies in dogs suggest variability in plasma imepitoin concentrations among individuals and sampling times. No correlation between plasma imepitoin concentration and seizure frequency was identified (Löscher et al., 2004). Although assays to measure plasma imepitoin concentrations were developed for the pharmacokinetic and clinical studies, these assays are not commercially available at present.
Efficacy
Imepitoin has demonstrated anticonvulsant activity in several seizure models in mice and rats including the maximal electroshock test, supramaximal stimulation with chemical convulsants such as pentylenetetrazol and bicuculline, audiogenic clonic seizures in rodent genetic models, and amygdala-kindled model of focal-onset seizures (Rostock et al., 1998a; Tober et al., 1998, 1999; Bialer et al., 2013).
In experimental dogs, imepitoin significantly increased the pentylenetetrazol-induced seizure threshold after single oral doses of 20 and 40 mg/kg and after 5-week-duration oral treatment with 5 or 40 mg/kg BID (Löscher et al., 2004).
In a recent study comparing the anticonvulsant efficacy of imepitoin and phenobarbital in the timed i.v. pentylenetetrazole (PTZ) seizure threshold test in dogs and, for comparison, in mice, imepitoin was moderately less potent than phenobarbital in increasing seizure threshold, but more tolerable in both species (Löscher et al., 2013).
The efficacy of imepitoin (in its initial formulation) as monotherapy has been investigated prospectively in 12 epileptic dogs (Löscher et al., 2004; Rieck et al., 2006). Imepitoin was started at 5 mg/kg every 12 h for 1 week and subsequently increased to 10 mg/kg every 12 h. If seizures persisted, the dosage was increased by up to 30 mg/kg every 12 h. The mean administered dosage was 20 mg/kg every 12 h. The efficacy of imepitoin was compared to retrospective data on efficacy of PB (mean daily dose 6 mg/kg, range 4–13 mg/kg) or PRM (mean daily dose 51 mg/kg, range 24–70 mg/kg) monotherapy in epileptic dogs (Table 22.2). On average dogs had experienced seizures for 5.1 ± 0.4 months prior to treatment initiation. Average duration of treatment was 7.7 ± 0.7 months for imepitoin,
5.9 ± 0.4 months for PB and 6.0 ± 0.6 months
Table 22.2. Efficacy of monotherapy with imepitoin, PB or PRM in epileptic dogs (modified from Löscher et al., 2004; Rieck et al., 2006).
Imepitoin PB PRM
| Number of dogs | 12 | 44 | 26 |
| Median seizure number per month before treatment | 1.6 | 1.71 | 1.75 |
| Median seizure number per month during treatment | 0.72 | 0.59a | 0.39 |
| Number (percentage) of dogs with >50% reduction in | 4/12 (33%) | 28/44 (64%) | 16/26 (62%) |
| seizure frequency during treatment | |||
| Number (percentage) of seizure-free dogs during | 1/12 (8%) | 9/44 (20%) | 4/26 (15%) |
| treatment | |||
| Number (percentage) of dogs with no decrease or an | 3/12 (25%) | 12/44 (27%) | 7/26 (27%) |
| increase in seizure frequency during treatment | |||
| Mean ± SD percentage of seizure reduction in dogs | 49.8 ± 11.3% | 72.4 ± 4.6% | 75.1 ± 5.1% |
| with decreased seizure frequency during treatment |
aSignificant difference in value before and during treatment.
for PRM. Although the percentage of dogs with no seizures or with >50% reduction in seizure frequency was higher in PB- or PRM-treated groups than in the imepitoin-treated group, this difference was not statistically significant. Seizure severity was subjectively described by the dog owners as decreased in 9/12 (75%) dogs treated with imepitoin, 24/44 (55%) dogs on PB and 10/26 (38%) dogs on PRM.
The efficacy of imepitoin (in its initial formulation) as adjunctive treatment to PB or PRM has been investigated prospectively in 17 epileptic dogs and compared to retrospective data on efficacy of KBr adjunctive therapy (Löscher et al., 2004; Rieck et al., 2006) (Table 22.3). Serum PB concentrations in these dogs ranged from 19.5 to 58.9 mg/ml (median 26.5, mean and standard deviation 32.0 ± 13.6 mg/ml). The imepitoin treatment protocol was the same as described above for monotherapy. Imepitoin average dosage was 17 mg/kg PO twice per day and average treatment duration was 5.6 ± 0.7 months. KBr was administered at 40–60 mg/kg PO per day for 7.3 ±
0.6 months. No significant difference was identified between the efficacy of imepitoin and KBr as adjunctive treatment. Seizure severity was subjectively described by the dog owners as decreased in 8/17 (47%) dogs treated with imepitoin and 4/12 (33%) dogs administered Br.
The efficacy of imepitoin in its final formulation was investigated in a multicentre, randomized, blinded study including 226 idiopathic epileptic dogs (SPC, 2013). The study included newly diagnosed idiopathic epileptic dogs with two or more generalized seizures during a 6-week retrospective baseline period preceding random assignment to the imepitoin (116 dogs) or PB (110 dogs) group. Imepitoin initial dosage was 10 mg/kg twice daily. Depending on clinical response this dosage could be increased, up to 30 mg/kg twice daily. The control group was administered PB at an initial dosage of 2 mg/kg twice daily. This dosage could also be increased, depending on clinical response up to 6 mg/kg twice daily, if required. Monthly seizure frequency during the retrospective baseline 6-week phase was compared to monthly seizure frequency during the prospective evaluation phase of 12 weeks. A total of 66 dogs in the imepitoin group and 88 dogs in the PB group were used in the efficacy evaluation analysis as it was necessary to exclude the remaining dogs for various reasons including non-idiopathic epilepsy diagnosed after study enrolment, owner compliance or withdrawal of owner consent and protocol deviations. The efficacy results are presented in Table 22.4.
Mean frequency of generalized seizures was reduced from 2.3 seizures per month in the imepitoin group and from 2.4 seizures per month in the PB group to approximately
1.1 seizures per month in both groups after 20 weeks of treatment. There was no significant difference in the mean seizure frequency per month during treatment (adjusted for baseline difference) between imepitoin and PB groups.
Table 22.3. Efficacy of imepitoin or KBr as adjunctive treatment to PB or PRM in epileptic dogs (modified from Löscher et al., 2004; Rieck et al., 2006).
Imepitoin KBr
Number of dogs 17 12 Median seizure number per month before adjunctive treatment 1.9 3.0 Median seizure number per month during adjunctive treatment 2.0 1.9 Number (percentage) of dogs with >50% reduction in seizure frequency 6/17 (35%) 5/12 (42%)
during adjunctive treatment Number of seizure-free dogs during adjunctive treatment 1/17 (6%) 0/12 (0%) Number of dogs with no decrease or an increase in seizure frequency 7/17 (41%) 5/12 (42%)
during adjunctive treatment Mean ± SD percentage of seizure reduction in dogs with decreased 47.2 ± 8.8% 59.7 ± 5.9% seizure frequency during adjunctive treatment
Imepitoin 501
Table 22.4. Efficacy of monotherapy with imepitoin or PB in idiopathic epileptic dogs (SPC, 2013).
Treatment group Imepitoin PB
| Number of dogs in efficacy analysis population | 66 | 88 |
|---|---|---|
| Mean monthly seizure frequency at baseline | 2.3 ± 1.58 | 2.4 ± 1.41 |
| Mean monthly seizure frequency during treatment | 1.07 ± 2.40 | 1.11 ± 3.21 |
| Number (percentage) of seizure-free dogs during treatment | 30/66 (45%) | 51/88 (58%) |
| Number (percentage) of dogs with ≥50% reduction in monthly | 50/66 (76%) | 73/88 (83%) |
| seizure frequency during treatment |
Imepitoin has also demonstrated anxiolytic activity in rodent models (Rostock et al., 1998b). The anxiolytic activity was not associated with the adverse reactions of full agonist benzodiazepines such as sedation, muscle relaxation and, upon repeated administration, development of tolerance.
Summary Recommendations
• Imepitoin is a partial benzodiazepine
receptor agonist with anticonvulsant and anxiolytic properties in rodents and dogs.
• Imepitoin is extensively metabolized prior to elimination and predominately excreted faecally.
• Imepitoin has been used in combination with phenobarbital in a small number of dogs and no harmful clinical interactions were observed.
• Further investigation is required on
imepitoin pharmacokinetic interactions.
• Imepitoin is generally well tolerated in dogs.
• There are no therapeutic monitoring
recommendations for imepitoin.
investigated.
References
Bialer, M., Johannessen, S.I., Kupferberg, H.J., Levy, R.H., Loiseau, P. and Perucca, E. (1999) Progress report on new antiepileptic drugs: a summary of the fourth Eilat conference (EILAT IV). Epilepsy Research 34, 1–41.
Bialer, M., Johannessen, S.I., Levy, R.H., Perucca, E., Tomson, T. and White, H.S. (2013) Progress report on new antiepileptic drugs: A summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Research 103(1), 2–30.
Löscher, W., Potschka, H., Rieck, S., Tipold, A. and Rundfeldt, C. (2004) Anticonvulsant efficacy of the low-affinity partial benzodiazepine receptor agonist ELB 138 in a dog seizure model and in epileptic dogs with spontaneously recurrent seizures. Epilepsia 45(10), 1228–1239.
Löscher, W., Hoffmann, K., Twele, F., Potschka, H. and Töllner, K. (2013) The novel antiepileptic drug imepitoin compares favourably to other GABA-mimetic drugs in a seizure threshold model in mice and dogs. Pharmacology Research 77, 39–46.
Rieck, S., Rundfeldt, C. and Tipold, A. (2006) Anticonvulsant activity and tolerance of ELB138 in dogs with epilepsy: a clinical pilot study. Veterinary Journal 172(1), 86–95.
Rostock, A., Tober, C., Dost, R., Rundfeldt, C., Bartsch, C., Egerland, U., Stark, B., Schupke, H., Kronbach, T., Lankau, H.-J., Unverferth, K. and Engel, J. (1998a) AWD-131–138. Drugs of the Future 23, 253–255.
Rostock, A., Tober, C., Dost, R. and Bartsch, R. (1998b) AWD 131–138 is a potential novel anxiolytic without sedation and amnesia: a comparison with diazepam and buspirone. Naunyn-Schmied. Archives of Pharmacology 358(Suppl. 1), R68.
Rundfeldt, C. and Löscher, W. (2013) The pharmacology of imepitoin: the first partial benzodiazepine receptor agonist developed for the treatment of epilepsy. CNS Drugs Dec 20. [Epub ahead of print]
Schneider, S. (2001) AWD 131–8: 13-week oral toxicity study after repeated administration in beagle dogs and subsequent 6- week recovery period. ASTA Medica AG Study report no. A-04101/3000917357.
Sigel, E., Baur, R., Netzer, R. and Rundfeldt, C. (1998) The antiepileptic drug AWD 131–138 stimulates different recombinant isoforms of the rat GABA(A) receptor through the benzodiazepine binding site. Neuroscience Letters 245, 85–88.
Tober, C., Rostock, A. and Bartsch, R. (1998) AWD 131–138: a derivative of a series of imidazolinones with anticonvulsant activity. Naunyn-Schmied. Archives of Pharmacology 358(Suppl. 1), R68.
Tober, C., Rostock, A., White, H.S., Wolf, H.H. and Bartsch, R. (1999) Anticonvulsant activity of AWD 131–138 in genetic animal models of epilepsy. Naunyn-Schmied. Archives of Pharmacology 359(Suppl. 3), R97.
Summary of Product Characteristics (SPC) (2013) Summary of product characteristics. Available at: http:// www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/veterinary/002543/ WC500140840.pdf
23 Pathophysiology and Management
of Cluster Seizures
Simon Platt BVM&S MRCVS Dipl. ACVIM (Neurology) Dipl. ECVN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
The temporal distribution of seizures is often as clinically relevant as overall seizure frequency. An animal that experiences one day of seizures per year may be considered fairly well controlled, but if the seizures occur in a flurry or cluster, this episode may result in a yearly visit to an emergency room or even hospital admission.
The terms ‘acute repetitive seizures’ (ARS), ‘cluster seizures’, ‘serial seizures’, ‘flurry seizures’, describe a condition characterized by multiple generalized tonic, clonic or tonic-clonic seizures or multiple focal seizures with or without secondary generalization in patients with epilepsy occurring over a relatively brief period of time, generally 24 h (Haut et al., 2005a, b; Haut, 2006). Acute intervention with benzodiazepines may abort such seizures and decrease adverse outcomes. While the number of seizures required and the period in which they occur have seldom been defined for ARS, many reports consider ARS to consist of three or more focal or generalized seizure episodes over a 24 h period (Haut et al., 2005a, b; Haut, 2006).
In humans with a seizure disorder, those with catastrophic epilepsies (CE) are most at risk for ARS. CE generally includes people with generalized cryptogenic (unknown) or symptomatic (structural) epilepsy and a portion of individuals with undetermined causes of epilepsy with both generalized and focal features. At the core of the issue of seizure clustering is the question of whether seizure episodes are random, or whether patterns can be predicted and possibly prevented. Thus, investigation of the clustering phenomenon yields insights into both specific mechanisms of clustering and more general concepts of seizure occurrence.
The Timing of Seizures
One of the unfortunate hallmarks of epilepsy is the unpredictability of seizures. The investigation of whether seizures are truly random events or events that follow identifiable patterns is not new. Attempts to comprehensively identify factors influencing seizure occurrence date back more than a century in humans. Menstrual cycle and time of day were features first considered; others followed by characterizing seizure patterns in large groups of patients with epilepsy, with particular emphasis on external features such as time of day, weather and season, as well as internal factors such as emotional state and gastrointestinal distress (Berlin and Yeager, 1951; Perry, 1954; Bandler et al., 1957; Ruhenstroth-Bauer et al., 1984; Jacono and Robertson, 1987; Bazan et al., 2005; Sanchez Fernandez et al., 2012). While some of these early associations are no longer
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
reported (e.g. constipation), many (e.g. early-morning clustering of seizures) remain widely accepted today (Sanchez Fernandez et al., 2012). In humans as well as dogs and cats, recent studies have attempted to look at the onset of seizures and the lunar cycle (Baxendale and Fisher, 2008; Browand-Stainback et al., 2011). Seizure counts in animals were compared among each of the eight individual lunar phases, among each of eight exact lunar phase dates and by percentage of lunar illumination (Browand-Stainback et al., 2011). However, no statistical significance was found in any of these comparisons excluding a relationship between the onset of epileptic seizures and the phases of the moon in animals. However, in humans, nocturnal luminance rather than the moon phase may influence the occurrence of epileptic seizures (Baxendale and Fisher, 2008).
The advent of continuous EEG monitoring for people in the 1960s enhanced the evaluation of the temporal occurrence of seizures (Stevens et al., 1969, 1971). Spike and seizure patterns in subjects undergoing continuous monitoring were analysed for periodicity and pattern. The concept of seizures as nonrandom events based on statistical analysis began to emerge; the observation that the occurrence of a seizure appeared to have a relationship with prior and subsequent interictal periods was reported, as was the previously noted influence of sleep–wake cycles (Stevens et al., 1971).
Despite many advances in the diagnosis, classification and treatment of epilepsy in the ensuing decades, little progress has been made toward addressing perhaps the most essential question in epilepsy: why does a seizure occur when it does? This investigation is rooted in basic mechanisms of epileptogenicity and seizure termination, combined with accurate clinical data regarding seizure occurrence and patterns.
Mechanisms of Seizure Clustering
Epilepsy is a unique disorder in that seizures are typically self-limited. Most seizures do not require acute intervention to terminate, and prolonged seizures such as status epilepticus (SE; see Chapter 24), requiring abortive therapies, tend to be the exception. Mechanisms of seizure termination are poorly understood, related to impaired neuronal receptor and ion channel function, and possibly hypoxia-related mitochondrial dysfunction (Doman and Pelligra, 2004). While a discussion of neuronal excitability is beyond the scope of this chapter, the concept of clustering implies either impaired termination or increased excitation, possibly due to secondary alterations from an initial seizure that promote a second attack, or an excess of seizure-promoting factors (Tauboll et al., 1991).
Seizures occurring within 8 h of a prior seizure have been reported to be significantly more likely to arise from the same focus than seizures more widely separated in time (Haut et al., 1997). These data directly suggested that at least in some settings, an ictal focus may be more excitable, or less inhibited, following a first seizure, leading to a seizure cluster. This demonstration supported a commonly used definition of a seizure cluster as three seizures per 24 h, derived from the fact that seizures within 8 h are not truly ‘independent’. It was similarly noted that seizures from a secondary focus tended to occur consecutively rather than randomly (Blum, 1994), although a subsequent study did not confirm this finding (Haut et al., 2002).
Defining Seizure Clustering
It is difficult to assign a specific definition to seizure clustering. At the simplest level, a seizure cluster is a closely grouped series of seizures. This approach defines clustering clinically, a strategy that is relatively easy to employ in clinical settings. Another approach is to consider clustering to be an increase over the patient’s typical seizure frequency. A more interesting question addresses the underlying temporal distribution of seizures: is seizure occurrence random, or can patterns, periodicity and deviations from randomness be identified? This approach identifies a cluster as a deviation from randomness. There is no definitive clinical definition for a cluster or
Pathophysiology and Management of Cluster Seizures
series of seizures. Studies examining clinically defined seizure clustering patterns have used varying empiric definitions, including: two to four seizures per 48 h (Caraballo et al., 2004); three seizures per 24 h (Yen et al., 2001; Haut et al., 2002); or two generalized tonicclonic or three complex focal seizures in 4 h (Rose et al., 2003). Nonspecific definitions, such as ‘those having several convulsions within a day or two’, have also been described. In a large randomized controlled trial of treatment for acute repetitive seizures in humans, the condition was defined as ‘multiple seizures occurring with a 24 h period for adults or 12 h period for children, with a pattern distinguishable from the usual seizure pattern’ (Dreifuss et al., 1998).
Seizure Periodicity
If seizures are random, then the occurrence of one seizure does not increase or decrease the likelihood of a subsequent one. However, there is ample evidence that many seizures are not ‘random’. As discussed below, cyclical patterns do exist in epilepsy, including circadian and catamenial patterns; furthermore, factors such as medication noncompliance and sleep deprivation are clear precipitants. Numerous human data demonstrate the nonrandom periodicity of seizures, on both an individual and a population basis (Almqvist, 1955; Binnie et al., 1984; Balish et al., 1991; Tauboll et al., 1991; Bauer and Burr, 2001). Two specific examples of factors related to seizure periodicity are time of day and menstrual phase. Daily or ultradian patterns for epileptiform discharges and seizures have been clearly demonstrated, highest in sleep (Stevens et al., 1969; Kellaway et al., 1980; Binnie et al., 1984; Balish et al., 1991), and particularly in patients with primary generalized epilepsy. A peak in seizure occurrence at midnight has been reported in people but such reports in dogs remain anecdotal (Kotagal and Yardi, 2008). Many human studies have demonstrated periodicity unrelated to these factors, in proportions ranging from 30% to 49% (Binnie et al., 1984; Balish et al., 1991; Bauer and Burr, 2001). Periodicities described include circadian, circaseptin (6–8 days), circavigintan (21 days) and ‘near monthly’ (Balish et al., 1991; Bauer and Burr, 2001).
Prevalence of Seizure Clustering
The prevalence of clustering varies widely between studies and definitions (3–76%; Milton et al., 1987; Balish et al., 1991; Tauboll et al., 1991; Bauer and Burr, 2001; Yen et al., 2001; Rose et al., 2003; Haut, 2006; Martinez et al., 2009). Many human studies are biased toward patients with intractable epilepsy, who are more likely to maintain prospective long-term diaries. The range likely reflects varying patient populations and sample size and, most significantly, a lack of uniformity in approaching the definition. Prevalence in the inpatient human setting tends to be 50% or higher of idiopathic epileptics, possibly reflecting a medication withdrawal effect.
Limited data exist documenting the prevalence of cluster seizures (CS) in veterinary medicine. A study of 407 dogs with idiopathic epilepsy found that 41% experienced cluster seizures at least once during their seizure history (Monteiro et al., 2012). A recent study performed in Border collies with confirmed idiopathic epilepsy revealed that CS occurred in 94% of the cases and SE in 53% (Hulsmeyer et al., 2010). More recently, 48% of Australian shepherd dogs diagnosed with idiopathic epilepsy below 5 years of age suffered from both CS and SE, 12% SE only and 20% CS only (Weissl et al., 2012).
Limited work has been done evaluating the prevalence of seizure clustering in cats. One study evaluating 125 cats with primary and secondary causes of seizure activity documented CS in 53% and 59% of cases, respectively (Pakozdy et al., 2010). There was no statistical prevalence difference between the causes; however, specific causes of clustering were not discussed in this study.
Risk Factors for Experiencing Seizure Clusters
Various studies have addressed risk factors for being a ‘clustered’ case. A commonly
considered risk factor in humans is epilepsy localization, with extratemporal epilepsy, particularly frontal lobe epilepsy, demonstrated to be associated with seizure clustering, although this is not present in all studies (Laskowitz et al., 1995; Manford et al., 1996; Fogarasi et al., 2001; Haut et al., 2005b). In descriptive reports of frontal lobe epilepsy in adults and children, seizures were described as ‘tending to cluster’ in 50% of patients (Laskowitz et al., 1995; Fogarasi et al., 2001). Aetiology may play a role; head trauma has been reported to be significantly associated with patient-reported history of seizure clustering independent of epilepsy localization in people although no such association was reported in a recent canine study of 259 dogs with head trauma (Laskowitz et al., 1995; Friedenberg et al., 2012).
Another risk factor for clustering in people appears to be seizure control. Patients with more intractable epilepsy appear to be at higher risk of experiencing seizure clustering (Balish et al., 1991; Haut et al., 2005b). However, in this group, the occurrence of seizure clusters defined clinically may be a random phenomenon of high seizure frequency. Gender has not been demonstrated to be associated with seizure clustering in humans (Tauboll et al., 1991; Bauer and Burr, 2001), despite the demonstration of hormonal influence on seizure occurrence. In dogs it appears that gender and neuter status are significantly associated with the occurrence of CS (Monteiro et al., 2012). Intact dogs were 1.4 times more likely than neutered dogs to suffer from CS and intact male dogs were also twice as likely as neutered males to suffer from CS in the most recent study (Monteiro et al., 2012). In the same study, female dogs had more frequent CS than males, with intact female dogs having more frequent CS than neutered female dogs. Experimentally, oestrogen enhances and progesterone diminishes neuronal excitability, whereas testosterone has less consistent effects (Rosciszewska et al., 1986). However, an attempt to extrapolate the occurrence of hormonal associated (catamenial) epilepsy may not be possible in canine patients due to the difference in the species’ sexual cycle. The benefit of neutering canine patients with idiopathic epilepsy has not been evaluated but a potential therapeutic role for this procedure may exist. Further studies are necessary to assess the effect of neutering on the seizure pattern of individual patients.
Age does not appear to be an independent risk factor for clustering in dogs and humans; longer duration of epilepsy does increase clustering risk in people; this is unknown in dogs (Bauer et al., 1992; Bauer and Burr, 2001; Monteiro et al., 2012).
When the association between breed and the occurrence of CS and or SE in dogs was evaluated recently, German shepherd dogs and boxers were found to be significantly more likely to suffer from CS (Monteiro et al., 2012). An earlier study, which included cases with epilepsy of various causes, reported English foxhounds, pug dogs, teacup poodles, Boston terriers and Lakeland terriers as having a higher frequency of occurrence of CS or SE compared to single seizure episodes. In this study 35.1% had symptomatic (structural) epilepsy (Bateman and Parent, 1999).
In veterinary medicine there has been a lack of association between CS and SE in dogs (Saito et al., 2001; Monteiro et al., 2012). In humans, a significant association between these two events has been found in cases of extratemporal epilepsy and head trauma (Haut et al., 2005b).
Risks and Implications of Seizure Clustering
Seizure clusters, while not as life threatening as SE, have a significant impact on patient health and well-being (Lombroso, 1989). Clusters frequently result in emergency room visits in humans and, if left untreated, have been reported to evolve into SE (Mitchell, 1996). Clusters have been related to a higher overall seizure frequency, medication-resistant epilepsy and a higher mortality than that seen in patients with epilepsy and without clusters. Seizure clusters are often treated at home with benzodiazepines, which are safe but not without adverse effects (Dreifuss et al., 1998; Mitchell et al., 1999). Furthermore, the treating veterinarian should be aware that the presence of clusters may indicate a poor prognosis of epilepsy and a higher risk of SE (Haut et al., 1999).
Pathophysiology and Management of Cluster Seizures
In a canine study, time to euthanasia due to CS was not associated with CS frequency or severity; however, euthanasia itself was associated with a high frequency of CS episodes (Monteiro et al., 2012). This suggests that CS frequency may be more important than the severity of each episode in the owner’s decision to elect for euthanasia. In a previous study where risk factors for the development of SE in dogs with IE were evaluated, death in 6 of 36 cases was attributed to poorly controlled epilepsy and survival was significantly negatively associated with the presence of SE (Saito et al., 2001). Mean lifespan of the SE group was 8.3 years compared to 11.3 years in dogs with no episodes of SE. In the Border collie study where dogs included suffered from either CS or SE or both, the median survival time was 2.07 years (Hulsmeyer et al., 2010). All epilepsy-related deaths were the result of euthanasia apart from two dogs, which died in SE. In the study of 407 dogs with IE, the median age at death was 5.17 years but in dogs whose death was epilepsy related, median age at death was significantly lower compared with dogs that died of other causes (Hulsmeyer et al., 2010; Monteiro et al., 2012).
Therapeutic Considerations
In humans, other than the use of benzodiazepines for clusters and occasionally acetozolamide for catamenial patterns, no specific therapy for seizure clustering exists and no preventive measures have been clearly identified (Lombroso, 1989; Mitchell, 1996; Dreifuss et al., 1998; Mitchell et al., 1999). While a rare ‘true’ clusterer may be instructed on the immediate use of rectal diazepam following a seizure, infrequent clusterers cannot identify when a first seizure will be single or the beginning of a cluster. Probability estimates based on past seizure histories are not feasible in the typical clinical setting, and using a clinical definition to identify and treat clusterers will result in false positives.
Prospective data are needed to clarify these issues, and a uniform definition of clustering would provide a firm basis for future studies. The emergence of seizure prediction modelling may both enhance our understanding of the pathophysiology of clustering and present opportunities for potential interventions to prevent seizure clusters.
Three broad options exist for the treatment of CS in veterinary patients, which are not mutually exclusive. These are: (i) improve long-term seizure control; (ii) administer short-acting treatment at the time of a cluster with the aim of reducing possible ‘roll on’ seizures in the next few minutes; and (iii) administer long-acting medication with the aim of reducing ‘roll on’ seizures over the subsequent few hours. Improving long-term seizure control is discussed in several other chapters within this volume, including Chapters 12–22).
Short-Acting Treatment at the Time of a Cluster Event
Rectal benzodiazepines
Benzodiazepines are the first-line therapy for the immediate treatment of seizures because they are fast-acting and effective (Boothe, 1998; Lowenstein and Alldredge, 1998; Treiman et al., 1998). On account of the fact that intravenous and intramuscular benzodiazepine administration is not an acceptable option for CS treatment in an out-of-hospital/at-home setting, rectal administration has been suggested as a useful alternative in emergency situations. It has been well established that absorption of lipid-soluble drugs by the membranes of the colon and rectum is rapid and complete (Powers et al., 1991). Diazepam can be administered into the rectum using plastic administrators such as teat infusion cannulas or tom-cat catheters with a water- soluble lubricant. The efficacy of rectally delivered diazepam depends on several factors, not least the time that it takes for the drug to reach the therapeutic concentration and exactly what the reference concentration is in an individual dog. In humans, it is estimated that the individual therapeutic concentration is approximately 150 to 300 ng/ml (Powers et al., 1991). If this is the case in dogs, several of the canine studies have demonstrated that this level can be reached well within 30 min, the time period before serious neurologic morbidity ensues (Powers et al., 1991). However, the actual therapeutic level in dogs has not been well documented and may be anywhere from 300 to 1500 ng/ml (Mealey and Boothe, 1995).
Rectal diazepam is currently the only out-of-hospital treatment that has been evaluated for treatment of cluster seizures in dogs (Podell, 1995). Dogs with a history of cluster seizures were evaluated; these dogs showed a significant decrease in the number of cluster seizure events in a 24-h period as well as a significant decrease in the total cost of emergency care. In pharmacokinetic studies,
0.5 mg/kg of rectally delivered diazepam produced a mean peak total benzodiazepine concentration of approximately 500 ng/ml in four healthy beagles (Papich and Alcorn, 1995). A separate study using six healthy mix-breed dogs found that the mean peak total concentration of benzodiazepines was 474 ng/ml with a dose of 1 mg/kg (Mealey and Boothe, 1995). However, in the case that the dog has been on chronic phenobarbital, a higher dose of diazepam may be necessary. This is because concurrent long-term oral phenobarbital administration reduces the serum level of benzodiazepines achieved following rectally delivered diazepam (Wagner et al., 1998). The mean peak concentrations of benzodiazepines after rectal administration of diazepam in six dogs fell from 629 ng/ml to 274 ng/ml when the dogs were given phenobarbital (2.5 mg/kg every 12 h for 30 days) (Wagner et al., 1998). This phenomenon is due to the increased activity of the hepatic microsomal enzyme system caused by phenobarbital administration.
In human studies, rectal administration of a parenteral solution of 0.5 mg/kg diazepam is approximately 80% effective in controlling prolonged seizures, usually within 15 min (Morton et al., 1997). A gel formulation of diazepam in a prefilled single-dose syringe for rectal administration (Diastat) is commercially available for the treatment of seizure clusters in children and adults and has been shown to be very effective (Fakhoury et al., 2007). When compared to IV lorazepam, diazepam rectal gel was typically administered more quickly and reliably reducing seizure duration, potential neuronal injury and other potential complications (Fitzgerald et al., 2003). Administration of a single dose of Diastat was significantly more effective than a placebo in reducing the number of seizures in a human randomized study (Cereghino et al., 1998). In addition, Diastat increased the probability that the patients would remain seizure-free for 12 h after the treatment compared with patients who received the placebo. Reported adverse effects of Diastat such as sedation, are consistent with the known safety profile of diazepam.
Canine studies have previously demonstrated the importance of the drug formulation and the vehicle used in determining the bioavailability of rectally delivered diazepam. Parenteral diazepam is compounded with 40% propylene glycol and 10% ethyl alcohol. This is the formulation that is commercially available; however, it is not approved for use as a rectally delivered drug. When the use of this formulation of diazepam solution was administered in high doses to rats, no significant gross or microscopic changes of the rectal mucosa were observed (Powers et al., 1991). This provides evidence of its local safety, however, a potential complication of the use of diazepam per rectum in dogs may be the development of a peri-rectal abscess, as has been recorded in one dog (Podell et al., 1998). This would seem to be a technique-associated problem rather than a direct reaction to the drug. This does emphasize the need for a complete review of the rectal administration protocol with the owner. The method of delivery requiring the use of a glass ampoule, needle and syringe for dispensing and tubing for administration, is awkward and potentially injurious to patients or caregiver and has a likely potential for dosing error. Preferably diazepam suppositories or liquid rectal formulations can be prepared/used for each individual case, with the help of compounding pharmacies if not commercially available or too expensive. This practice may also reduce the concerns of scheduled drug misuse and abuse.
Other benzodiazepines have been evaluated pharmacokinetically following rectal administration. Pilot studies on the use of
0.2 mg/kg IV and 0.5–1.0 mg/kg rectal lorazepam in normal dogs suggested that lorazepam may not be acceptable for rectal
Pathophysiology and Management of Cluster Seizures
administration because of an extensive first pass effect (Lehmann and Boulieu, 1995). However, lorazepam can reach therapeutic blood concentrations when administered IV (Lehmann and Boulieu, 1995). Rectal administration of midazolam in humans has been shown to be safe and effective, with peak plasma concentrations occurring within 10 min after administration (Nordt and Clark, 1997). After rectal instillation of a solution of midazolam hydrochloride (0.3 mg/kg) in healthy human volunteers, a mean bioavailability of 52% was found (van Hoogdalem et al., 1991). Clinical effect, measured in terms of sedation and EEG activity, was seen to be comparable to that seen with IV administration. Rectal administration of the parenteral midazolam solution also achieved concentrations that would be considered therapeutic in people; however the Tmax was prolonged (39 ± 14.49 min). The bioavailability of 49% is improved from the previous report of almost no absorption (Court and Greenblatt, 1992). Pharmacokinetic reports of rectal midazolam in humans are variable with bioavailability ranging from 18% to 52% and a Tmax of 12.1–31 min (Clausen et al., 1988; Payne et al., 1989; Malinovsky et al., 1993, 1995). Rectal diazepam in dogs has been shown to have a bioavailability from 51.7% (± 21.8%) to 79.9% (± 20.7%) and a Tmax of 14.3 min (± 3.7) (Mealey and Boothe, 1995; Papich and Alcorn, 1995). Based on these pharmacokinetics, rectal diazepam would seem to be superior to rectal midazolam as an anticonvulsant. Rectal administration of
0.2 mg/kg midazolam in healthy dogs resulted in erratic systemic availability with undetectable to low plasma concentrations with the conclusion from this study being that rectal administration of midazolam is likely to be of limited efficacy for treating seizures in dogs (Schwartz et al., 2012)
Nasal benzodiazepines
The intranasal route has been investigated extensively in humans and has been found not only to be more convenient and socially acceptable than rectal diazepam, but also yielded equal or better results in regards to anti-epileptic activity and onset of action (Fisgin et al., 2002; Harbord et al., 2004; Bhattacharyya et al., 2006; Ivaturi et al., 2013). Certain nasal preparations of midazolam have even demonstrated higher plasma concentrations in a shorter time than an equivalent dose administered IM (Wermeling et al., 2006). The effectiveness of intranasal midazolam is such that it has been recommended in consensus guidelines as an alternative for the rapid treatment of humans with SE (Harbord et al., 2004; Meierkord et al., 2006; Prasad et al., 2007).
Diazepam administered intranasally (IN) to dogs has been shown to reach total benzodiazepine plasma concentrations within the human anticonvulsant ranges (Mealey and Boothe, 1995; Papich and Alcorn, 1995; Platt et al., 2000). Intranasal administration of midazolam, triazolam and flurazepam yielded maximum plasma concentration within 15 min in dogs, but the dose given was not reported (Lui et al., 1991). Another pharmacokinetic study of intranasal midazolam administered at 1.5 mg/kg found a bioavailability of only 10% (Henry et al., 1998).
Although intranasal delivery of the parenteral formulation has been proven effective for treatment of SE in humans, there are some drawbacks. The most significant problem reported is the large volume needed to ensure adequate dosing. The large volume results in run-off out of the front of the nose and/or down the nasopharynx to be ingested, decreasing the bioavailability and efficacy (Burstein et al., 1997; Wermeling, 2009). The limitations encountered with administration of a large volume prompted a number of studies evaluating novel, highly concentrated formulations of midazolam (Gudmundsdottir et al., 2001; Knoester et al., 2002; Wermeling et al., 2006, 2009). Investigations into alternative formulations have yielded more rapid and complete absorption than the commercially available 5 mg/ml product (Gudmundsdottir et al., 2001; Loftsson et al., 2001; Knoester et al., 2002; Dale et al., 2006; Wermeling et al., 2006, 2009). However, there are limited data regarding the use of any of these alternative formulations in a clinical setting.
The dose used for most studies reporting successful treatment of seizures with intranasal midazolam (parenteral solution) in humans is 0.2 mg/kg, a dose that is derived from prior sedation/anaesthesia studies (O’Regan et al., 1996; Lahat et al., 2000; Scheepers et al., 2000; Fisgin et al., 2002; Harbord et al., 2004; Mahmoudian and Zadeh, 2004; Wilson et al., 2004; Bhattacharyya et al., 2006; Holsti et al., 2007). Pharmacokinetic studies in humans evaluating intranasal administration of the parenteral 5 mg/ml formulation at doses of
0.2 mg/kg have demonstrated mean peak plasma concentrations between 104 ng/ml and 182 ng/ml within 12 min, well above the 40 ng/ml considered to be the minimum therapeutic level for sedation in adults (Allonen et al., 1981; Rey et al., 1991; Malinovsky et al., 1993). Estimated bioavailability of the parenteral 5 mg/ml formulation administered at
0.2 mg/kg IN in humans ranges from 50 to 83% (Rey et al., 1991; Burstein et al., 1997; Knoester et al., 2002). Intranasal midazolam at
0.2 mg/kg has been demonstrated to penetrate the human brain in 2–5 min, as shown by the appearance of beta activity in the EEG and suppressed epileptic activity (O’Regan et al., 1996). This suggests that early entry of modest midazolam concentrations into the brain compartment has anti-epileptic effect and that the plasma midazolam concentration needed to stop a seizure could be far less than the concentration needed to produce sedation (Wermeling et al., 2009).
A recent canine study evaluating a novel 0.4% hydroxypropyl methylcellulose midazolam gel showed it to be readily absorbed when administered IN with a pharmacokinetic profile superior than the parenteral solution given IN or rectally to dogs (Eagleson et al., 2012). More specifically, the 0–30 min area under the curve following IN gel administration was significantly higher than IN solution and rectal administration (p <0.001), which is a key parameter when evaluating a drug to be used in a setting where time to clinical effect is important. The mean peak plasma concentration of 450 ng/ml seen is more than twice the peak plasma concentrations seen in human studies using the parenteral solution at the same dose via IN route. The T and C of the intranasal gel are
maxmax
comparable to IM administration of a 0.5 mg/kg dose in dogs (C= 549 ± 121 ng/ml; T=
max max
8 ± 2 min) (Court and Greenblatt, 1992).
Alternative intranasal formulations of midazolam have demonstrated favourable pharmacokinetics in humans. The common feature to all the experimental formulations is a solubility enhancer, resulting in a more concentrated product allowing delivery of a much smaller volume. The aim is to generate a product that could deliver an effective dose in 0.1–0.2 ml, an accepted retention volume in the human nose (Romeo et al., 1998). Due to the extreme phenotypical variation seen in the domestic dog (in both size of animal and conformation of the head), an average nasal retention volume in dogs is difficult to define. None the less, a highly concentrated/low volume product would be advantageous in brachycephalic and large breed dogs. All pharmacokinetic studies in humans evaluating alternative midazolam formulations have used doses less than 0.1 mg/kg. The highest peak plasma concentration reported in these studies is 80 ng/ml reached within 7.2 or 10.3 min (Wermeling et al., 2006; Haschke et al., 2010). Reported bioavailabilities of the alternative formulations have ranged anywhere from 64 to 92% (Gudmundsdottir et al., 2001; Haschke et al., 2010).
Pharmacokinetics of IN delivered drugs can be further improved using bioadhesive gels that prolong contact time with the nasal mucosa and metered spray delivery systems which deposit medication more rostrally in the nasal antrum so that clearance from the nasopharynx occurs more slowly (Harris et al., 1986; Kohler et al., 1986; Morimoto et al., 1987; Henry et al., 1998; Najafabadi et al., 2004). The methylcellulose used in the recent canine study is a viscosity-enhancing agent that can promote retention in the nasal cavity by slowing the ciliary movement of mucus (Wermeling, 2009; Eagleson et al., 2012). Nasal spray delivery systems are used in the majority of the studies in humans. In dogs, the use of an atomizer has been shown to improve the pharmacokinetics of intranasal midazolam in one study but made no difference in another (Henry et al., 1998; Musulin et al., 2011).
In most humans, transient nasal irritation, tearing and a raw throat sensation are reported in pharmacokinetic studies using both the parenteral solution and alternative formulations. Midazolam is water soluble;
Pathophysiology and Management of Cluster Seizures
however, to remain in solution it must be buffered to approximately pH 3. It is thought that the low pH is the cause of most of the discomfort reported; however, alternative formulations with a pH of 4–4.2 also caused the same discomfort, suggesting it may be the midazolam itself that is the irritant (Gudmundsdottir et al., 2001; Knoester et al., 2002). The significance of the aforementioned side effects, however, is questionable as the majority of patients treated for a seizure with intranasal midazolam do not report any of them (de Haan et al., 2010; Wermeling et al., 2009).
It should be noted that IN administration might carry a higher risk of injury (due to bite injuries) to the caregiver than that posed by rectal administration. At this time, the aforementioned risk and lack of a proprietary formulation makes IN administration less favourable than it is in human SE treatment.
Long-Acting Treatment at the Time of a Cluster Event
The following options should be viewed as at-home therapies for those animals that are known to exhibit cluster seizures. The treatments are not intended as a method to avoid veterinary care and counselling but offer an ‘on the spot’ therapy that may prevent cluster seizures from occurring or at least reduce the amount of seizures exhibited within a 24 h period. As such, the owners can be advised to attempt one of the below options either at the time of a prodrome, if reliable detection is possible, or at least at the time of the first seizure.
Oral clorazepate
Clorazepate (clorazepate dipotassium) is a benzodiazepine pro-drug that acts by enhancing GABA activity in the brain. Nordiazepam is the active metabolite produced following rapid hydrolysation, which occurs in the stomach when the drug is administered orally. The serum elimination half-life of nor-diazepam after clorazepate administration is 4–6 h in dogs but may be as short as 3 h. After oral administration of 2 mg/kg, clorazepate reaches peak benzodiazepine concentration of 446 to 1542 ng/ml in dogs; mean residence time is 8.5 h but this increases with multiple administrations to approximately 12 h (Forrester et al., 1990). Oral doses as high as 2 mg/kg can result in profound sedation and ataxia but such signs may resolve 3 to 4 days after treatment (Scherkl et al., 1989). Tolerance to the use of this drug has been described in some dogs with resultant reduction in its anticonvulsant properties (Scherkl et al., 1989). In essence this drug is a short-term anticonvulsant that can be effective when administered orally.
Anecdotally, this drug can be given as a pulsed therapy adjunctively to the maintenance medication which the animal is on, beginning at the lower end of the recommended dose (0.5–2.0 mg/kg PO q8h). The success of this approach may depend on the tolerance of the owner to the ensuing sedation and the ability to predict a cluster based on the first seizure event. There are no data on the use of this approach but it is potentially very effective and cheap. The duration of treatment can be short (1–3 days).
The author does not recommend the use of this drug in dogs receiving phenobarbital or in cats unless absolutely necessary. Administration of phenobarbital alters the disposition of clorazepate such that the amount of nordiazepam in circulation during each dose interval is significantly reduced. Adequate control of seizures in epileptic dogs, therefore, may require higher dosages of clorazepate when it is co-administered with phenobarbital (Forrester et al., 1993).
Oral levetiracetam
The mechanism of action of levetiracetam (LEV) is unique. It does not appear to work directly through GABA-ergic facilitation, inhibition of Na+ channels, or modulation of low-voltage activated Ca2+ currents (see Chapter 16) (Lynch et al., 2004). Instead, its binding site in the brain appears to be a specific synaptic vesicle protein (SVA2) involved in the modulation of neurotransmitter release, reuptake and recycling (Lynch et al., 2004; Volk et al., 2008). Approximately one-third of the parent drug undergoes renal excretion related to glomerular filtration rate. Active drug is also hydrolysed by enzymes in a variety of tissues (Benedetti et al., 2004). In dogs with seizures resistant to PB and KBr therapy, approximately 60% of dogs had ³50% reduction in seizure frequency when LEV was given at a dosage of 10–20 mg/kg orally three times daily (Volk et al., 2008). Although the therapeutic serum target concentration of LEV in dogs is unknown it is likely similar to that in humans (5 to 45 mg/ml). Reported adverse effects attributed to LEV in dogs and cats appear infrequent and include sedation, ataxia, decreased appetite and vomiting in dogs and transient mild lethargy and inappetence in cats (Volk et al., 2008; Muñana et al., 2012). Dose reduction or permanent discontinuation has been necessary in a few cases to resolve these adverse effects. Transient mild to moderate hypersalivation has been reported in cats following administration of the commercially available LEV oral suspension (Patterson et al., 2008; Volk et al., 2008; Carnes et al., 2011). Importantly, the pharmacokinetics appear favourable for oral administration during CS. The bioavailability of oral administration is nearly 100% and the maximum serum concentration following intramuscular administration is attained in 40 min (Patterson et al., 2008). Moreover, the T1/2 in dogs is short, ranging from 2.3 to 4 h (Isoherranen et al., 2001; Benedetti et al., 2004; Dewey et al., 2008; Patterson et al., 2008). Subcutaneous (SC) administration of LEV at 60 mg/kg results in plasma LEV concentration above the proposed human reference range of 5–46 mg/ml at all time-points (see Chapter 16) (Leppik, 2002). However, the use of an injectable medication at home by owners may be considered a risk to the owner and to the patient.
The main disadvantage to the use of LEV has been expense and the possibility that concurrent use of phenobarbital may reduce its serum levels. However, recently a generic formulation has been developed which is comparable to the cost of phenobarbital or parenteral formulations of benzodiazepines. Preliminary data from the author’s work on the extended release formulation of LEV not only corroborate the established T1/2 of the standard release formulation in dogs but importantly provides meaningful data documenting rapid achievement of peak concentrations and maintenance of serum concentrations well above the accepted serum reference range (5 to 45 mg/ml).
Again anecdotally, the use of a pulsed oral dosing regimen of LEV, especially the extended release formulation, has been suggested as an approach to the treatment of at-home seizure clusters. If the animal is already on phenobarbital, a higher dose of LEV is advised (at least 20 mg/kg rather than 10 mg/kg). Similar to the use of clorazepate, in this situation, the success of this approach may depend on the prediction of cluster activity but in this drug’s case, there will be limited sedation and liver metabolism is not a concern. The use of IV LEV has been documented as a successful treatment of acute repetitive seizures in humans, experimental animal models and clinical canine cases (Abend et al., 2009a, b; Modur et al., 2010; Hardy et al., 2012); this would be a useful in-hospital option for veterinary patients regardless of the maintenance therapy that is being used. A randomized, placebo-controlled, double-masked study including 19 dogs with SE or cluster seizures (acute repetitive seizures) has shown that administration of intravenous LEV (30 or 60 mg/kg) in addition to diazepam resulted in a significantly higher responder rate (56% after LEV) compared to placebo and diazepam (10%). In addition, dogs in the placebo group required significantly more boluses of diazepam compared with the LEV group (Hardy et al., 2012). Further studies are required to investigate LEV efficacy in SE and cluster seizures in a larger number of dogs and in cats.
Vagal nerve stimulation
Vagal nerve stimulation (VNS) has been used as an adjunctive maintenance treatment for chronic seizures. The modern VNS system includes a pulse generator that resides in the chest that connects to the left vagus nerve via an enclosed wire tunnelled under the skin. Asmall coil, through which current is induced, wraps around the vagus nerve. Afferent stimulation is sent to the brain. This stimulation is proposed to reduce the incidence and severity of seizures. Although the device provides
Pathophysiology and Management of Cluster Seizures
cyclic stimulation 24 h daily on an adjustable on-off cycle, the device can be triggered by the patient or the caregiver to give an additional, usually more intense, stimulation (Poukas et al., 2011). Extra (rescue) stimulation is triggered by swiping a wrist-magnet, provided with the device, across their chest, ‘the magnet-mode’. These stimulations are longer and more intense (more current is delivered) than those provided at baseline. The magnet can be used throughout a clinical event. As this therapy remains prohibitively expensive in veterinary medicine for most people, its utility as an option for clusters seems very limited. However, a new handheld vagal nerve stimulator is currently being trialled as an intermittent treatment in those dogs that exhibit acute repetitive seizures or clusters. The unit is commercially available, affordable, available for the treatment of migraines in humans and easy to use. Further information about this modality is provided in Chapter 25.
Summary
The phenomenon of seizure clustering is clinically recognized and particularly prevalent in certain settings, although patients who experience all their seizures in clusters appear to be rare. Clustering may be defined clinically as a closely grouped series of seizures or, statistically, as the occurrence of seizures deviating from an expected random distribution. Seizures in clusters have demonstrated dependence, in that the presence of a seizure event influences the probability of a subsequent one. Patients with toxicity, metabolic disease, a history of head trauma, intractable epilepsy and long epilepsy duration may be at particular risk for seizure clusters. Among the implications of seizure clustering are concerns for patient safety. Prospective data are needed to better identify risks and precipitants of clustering, in an effort to identify preventive measures and enhance our understanding of the pathophysiology of seizure patterns and occurrence.
References
Abend, N.S., Florance, N., Finkel, R.S., Licht, D.J. and Dlugos, D.J. (2009a) Intravenous levetiracetam terminates refractory focal status epilepticus. Neurocritical Care 10, 83–86.
Abend, N.S., Monk, H.M., Licht, D.J. and Dlugos, D.J. (2009b) Intravenous levetiracetam in critically ill children with status epilepticus or acute repetitive seizures. Pediatric Critical Care Medicine 10, 505–510.
Allonen, H., Ziegler, G. and Klotz, U. (1981) Midazolam kinetics. Clinical Pharmacology and Therapeutics 30, 653–661.
Almqvist, R. (1955) The rhythm of epileptic attacks and its relationship to the menstrual cycle. Acta Psychiatria Neurologia Scandinavica Suppl. 105, 1–116.
Balish, M., Albert, P.S. and Theodore, W.H. (1991) Seizure frequency in intractable partial epilepsy: a statistical analysis. Epilepsia 32, 642–649.
Bandler, B., Dykens, J.W., Kaufman, I.C., Schleifer, M. and Shapiro, L.N. (1957) Seizures and the menstrual cycle. American Journal of Psychiatry 113, 704–708.
Bateman, S.W. and Parent, J.M. (1999) Clinical findings, treatment, and outcome of dogs with status epilepticus or cluster seizures: 156 cases (1990–1995). Journal of the American Veterinary Medical Association 215, 1463–1468.
Bauer, J. and Burr, W. (2001) Course of chronic focal epilepsy resistant to anticonvulsant treatment. Seizure 10, 239–246.
Bauer, J., Stefan, H., Schrell, U., Uhlig, B., Landgraf, S., Neubauer, U., Neundorfer, B. and Burr, W. (1992) Serum prolactin concentrations and epilepsy. A study which compares healthy subjects with a group of patients in presurgical evaluation and circadian variations with those related to seizures. European Archives of Psychiatry and Clinical Neuroscience 241, 365–371.
Baxendale, S. and Fisher, J. (2008) Moonstruck? The effect of the lunar cycle on seizures. Epilepsy and Behavior 13, 549–550.
Bazan, A.C., Montenegro, M.A., Cendes, F., Min, L.L. and Guerreiro, C.A. (2005) Menstrual cycle worsening of epileptic seizures in women with symptomatic focal epilepsy. Arquivos de Neuro-psiquiatr 63, 751–756.
Benedetti, M.S., Coupez, R., Whomsley, R., Nicolas, J.M., Collart, P. and Baltes, E. (2004) Comparative pharmacokinetics and metabolism of levetiracetam, a new anti-epileptic agent, in mouse, rat, rabbit and dog. Xenobiotica 34, 281–300.
Berlin, I.N. and Yeager, C.L. (1951) Correlation of epileptic seizures, electroencephalograms and emotional state; some preliminary observations in several children. AMA American Journal of Diseases of Children 81, 664–670.
Bhattacharyya, M., Kalra, V. and Gulati, S. (2006) Intranasal midazolam vs rectal diazepam in acute childhood seizures. Pediatric Neurology 34, 355–359.
Binnie, C.D., Aarts, J.H., Houtkooper, M.A., Laxminarayan, R., Martins Da Silva, A., Meinardi, H., Nagelkerke, N. and Overweg, J. (1984) Temporal characteristics of seizures and epileptiform discharges. Electroencephalography and Clinical Neurophysiology 58, 498–505.
Blum, D. (1994) Prevalence of bilateral partial seizure foci and implications for electroencephalographic telemetry monitoring and epilepsy surgery. Electroencephalography and Clinical Neurophysiology 91, 329–336.
Boothe, D.M. (1998) Anticonvulsant therapy in small animals. Veterinary Clinics of North America - Small Animal Practice 28, 411–448.
Browand-Stainback, L., Levesque, D. and Mcbee, M. (2011) Canine and feline epileptic seizures and the lunar cycle: 2,507 seizures (2000–2008). Journal of the American Animal Hospital Association 47, 324–328.
Burstein, A.H., Modica, R., Hatton, M., Forrest, A. and Gengo, F.M. (1997) Pharmacokinetics and pharmacodynamics of midazolam after intranasal administration. Journal of Clinical Pharmacology 37, 711–718.
Caraballo, R.H., Cersosimo, R.O. and Fejerman, N. (2004) Benign focal seizures of adolescence: a prospective study. Epilepsia 45, 1600–1603.
Carnes, M.B., Axlund, T.W. and Boothe, D.M. (2011) Pharmacokinetics of levetiracetam after oral and intravenous administration of a single dose to clinically normal cats. American Journal of Veterinary Research 72, 1247–1252.
Cereghino, J.J., Mitchell, W.G., Murphy, J., Kriel, R.L., Rosenfeld, W.E. and Trevathan, E. (1998) Treating repetitive seizures with a rectal diazepam formulation: a randomized study. The North American Diastat Study Group. Neurology 51, 1274–1282.
Clausen, T.G., Wolff, J., Hansen, P.B., Larsen, F., Rasmussen, S.N., Dixon, J.S. and Crevoisier, C. (1988) Pharmacokinetics of midazolam and alpha-hydroxy-midazolam following rectal and intravenous administration. British Journal of Clinical Pharmacology 25, 457–463.
Court, M.H. and Greenblatt, D.J. (1992) Pharmacokinetics and preliminary observations of behavioral changes following administration of midazolam to dogs. Journal of Veterinary Pharmacology and Therapeutics 15, 343–350.
Dale, O., Nilsen, T., Loftsson, T., Hjorth Tonnesen, H., Klepstad, P., Kaasa, S., Holand, T. and Djupesland, P.G. (2006) Intranasal midazolam: a comparison of two delivery devices in human volunteers. Journal of Pharmacy and Pharmacology 58, 1311–1318.
De Haan, G.J., Van Der Geest, P., Doelman, G., Bertram, E. and Edelbroek, P.A (2010) A comparison of midazolam nasal spray and diazepam rectal solution for the residential treatment of seizure exacerbations. Epilepsia 51, 478–482.
Dewey, C.W., Bailey, K.S., Boothe, D.M., Badgley, B.L. and Cruz-Espindola, C. (2008) Pharmacokinetics of single-dose intravenous levetiracetam administration in normal dogs. Journal of Veterinary Emergency and Critical Care 18, 153–157.
Doman, G. and Pelligra, R. (2004) A unifying concept of seizure onset and termination. Med Hypotheses 62, 740–745.
Dreifuss, F.E., Rosman, N.P., Cloyd, J.C., Pellock, J.M., Kuzniecky, R.I., Lo, W.D., Matsuo, F., Sharp, G.B., Conry, J.A., Bergen, D.C. and Bell, W.E. (1998) A comparison of rectal diazepam gel and placebo for acute repetitive seizures. New England Journal of Medicine 338, 1869–1875.
Eagleson, J.S., Platt, S.R., Strong, D.L., Kent, M., Freeman, A.C., Nghiem, P.P., Zheng, B. and White, C.A. (2012) Bioavailability of a novel midazolam gel after intranasal administration in dogs. American Journal of Veterinary Research 73, 539–545.
Fakhoury, T., Chumley, A. and Bensalem-Owen, M. (2007) Effectiveness of diazepam rectal gel in adults with acute repetitive seizures and prolonged seizures: a single-center experience. Epilepsy and Behavior 11, 357–360.
Fisgin, T., Gurer, Y., Tezic, T., Senbil, N., Zorlu, P., Okuyaz, C. and Akgun, D. (2002) Effects of intranasal midazolam and rectal diazepam on acute convulsions in children: prospective randomized study. Journal of Child Neurology 17, 123–126.
Pathophysiology and Management of Cluster Seizures
Fitzgerald, B.J., Okos, A.J. and Miller, J.W. (2003) Treatment of out-of-hospital status epilepticus with diazepam rectal gel. Seizure 12, 52–55.
Fogarasi, A., Janszky, J., Faveret, E., Pieper, T. and Tuxhorn, I. (2001) A detailed analysis of frontal lobe seizure semiology in children younger than 7 years. Epilepsia 42, 80–85.
Forrester, S.D., Brown, S.A., Lees, G.E. and Hartsfield, S.M. (1990) Disposition of clorazepate in dogs after single- and multiple-dose oral administration. American Journal of Veterinary Research 51, 2001–2005.
Forrester, S.D., Wilcke, J.R., Jacobson, J.D. and Dyer, K.R. (1993) Effects of a 44-day administration of phenobarbital on disposition of clorazepate in dogs. American Journal of Veterinary Research 54, 1136–1138.
Friedenberg, S.G., Butler, A.L., Wei, L., Moore, S.A. and Cooper, E.S. (2012) Seizures following head trauma in dogs: 259 cases (1999–2009). Journal of the American Veterinary Medical Association 241, 1479–1483.
Gudmundsdottir, H., Sigurjonsdottir, J.F., Masson, M., Fjalldal, O., Stefansson, E. and Loftsson, T. (2001) Intranasal administration of midazolam in a cyclodextrin based formulation: bioavailability and clinical evaluation in humans. Pharmazie 56, 963–966.
Harbord, M.G., Kyrkou, N.E., Kyrkou, M.R., Kay, D. and Coulthard, K.P. (2004) Use of intranasal midazolam to treat acute seizures in paediatric community settings. Journal of Paediatrics and Child Health 40, 556–558.
Hardy, B.T., Patterson, E.E., Cloyd, J.M., Hardy, R.M. and Leppik, I.E. (2012) Double-masked, placebo-controlled study of intravenous levetiracetam for the treatment of status epilepticus and acute repetitive seizures in dogs. Journal of Veterinary Internal Medicine 26, 334–340.
Harris, A.S., Nilsson, I.M., Wagner, Z.G. and Alkner, U. (1986) Intranasal administration of peptides: nasal deposition, biological response, and absorption of desmopressin. Journal of Pharmacological Science 75, 1085–1088.
Haschke, M., Suter, K., Hofmann, S., Witschi, R., Frohlich, J., Imanidis, G., Drewe, J., Briellmann, T.A., Dussy, F.E., Krahenbuhl, S. and Surber, C. (2010) Pharmacokinetics and pharmacodynamics of nasally delivered midazolam. British Journal of Clinical Pharmacology 69, 607–616.
Haut, S.R. (2006) Seizure clustering. Epilepsy and Behavior 8, 50–55.
Haut, S.R., Legatt, A.D., O’Dell, C., Moshe, S.L. and Shinnar, S. (1997) Seizure lateralization during EEG monitoring in patients with bilateral foci: the cluster effect. Epilepsia 38, 937–940.
Haut, S.R., Shinnar, S., Moshe, S.L., O’Dell, C. and Legatt, A.D. (1999) The association between seizure clustering and convulsive status epilepticus in patients with intractable complex partial seizures. Epilepsia 40, 1832–1834.
Haut, S.R., Swick, C., Freeman, K. and Spencer, S. (2002) Seizure clustering during epilepsy monitoring. Epilepsia 43, 711–715.
Haut, S.R., Lipton, R.B., Levalley, A.J., Hall, C.B. and Shinnar, S. (2005a) Identifying seizure clusters in patients with epilepsy. Neurology 65, 1313–1315.
Haut, S.R., Shinnar, S. and Moshe, S.L. (2005b) Seizure clustering: risks and outcomes. Epilepsia 46, 146–149.
Henry, R.J., Ruano, N., Casto, D. and Wolf, R.H. (1998) A pharmacokinetic study of midazolam in dogs: nasal drop vs. atomizer administration. Pediatric Dentistry 20, 321–326.
Holsti, M., Sill, B.L., Firth, S.D., Filloux, F.M., Joyce, S.M. and Furnival, R.A. (2007) Prehospital intranasal midazolam for the treatment of pediatric seizures. Pediatric Emergency Care 23, 148–153.
Hulsmeyer, V., Zimmermann, R., Brauer, C., Sauter-Louis, C. and Fischer, A. (2010) Epilepsy in Border Collies: clinical manifestation, outcome, and mode of inheritance. Journal of Veterinary Internal Medicine 24, 171–178.
Isoherranen, N., Yagen, B., Soback, S., Roeder, M., Schurig, V. and Bialer, M. (2001) Pharmacokinetics of levetiracetam and its enantiomer (R)-alpha-ethyl-2-oxo-pyrrolidine acetamide in dogs. Epilepsia 42, 825–830.
Ivaturi, V., Kriel, R., Brundage, R., Loewen, G., Mansbach, H. and Cloyd, J. (2013) Bioavailability of intranasal vs. rectal diazepam. Epilepsy Research 103, 254–261.
Jacono, J.J. and Robertson, J.M. (1987) The effects of estrogen, progesterone, and ionized calcium on seizures during the menstrual cycle of epileptic women. Epilepsia 28, 571–577.
Kellaway, P., Frost, J.D., Jr and Crawley, J.W. (1980) Time modulation of spike-and-wave activity in generalized epilepsy. Annals of Neurology 8, 491–500.
Knoester, P.D., Jonker, D.M., Van Der Hoeven, R.T., Vermeij, T.A., Edelbroek, P.M., Brekelmans, G.J. and De Haan, G.J. (2002) Pharmacokinetics and pharmacodynamics of midazolam administered as a concentrated intranasal spray. A study in healthy volunteers. British Journal of Clinical Pharmacology 53, 501–507.
Kohler, M., Hellstern, P., Miyashita, C., Von Blohn, G. and Wenzel, E. (1986) Comparative study of intranasal, subcutaneous and intravenous administration of desamino-D-arginine vasopressin (DDAVP). Thrombosis and Haemostasis 55, 108–111.
Kotagal, P. and Yardi, N. (2008) The relationship between sleep and epilepsy. Seminars in Pediatric Neurology 15, 42–49.
Lahat, E., Goldman, M., Barr, J., Bistritzer, T. and Berkovitch, M. (2000) Comparison of intranasal midazolam with intravenous diazepam for treating febrile seizures in children: prospective randomised study. British Medical Journal 321, 83–86.
Laskowitz, D.T., Sperling, M.R., French, J.A. and O’Connor, M.J. (1995) The syndrome of frontal lobe epilepsy: characteristics and surgical management. Neurology 45, 780–787.
Lehmann, B. and Boulieu, R. (1995) Determination of midazolam and its unconjugated 1-hydroxy metabolite in human plasma by high-performance liquid chromatography. Journal of Chromatography B - Biomedical Applications 674, 138–142.
Leppik, I.E. (2002) Three new drugs for epilepsy: levetiracetam, oxcarbazepine, and zonisamide. Journal of Child Neurology 17(Suppl. 1), S53–57.
Loftsson, T., Gudmundsdottir, H., Sigurjonsdottir, J.F., Sigurdsson, H.H., Sigfusson, S.D., Masson, M. and Stefansson, E. (2001) Cyclodextrin solubilization of benzodiazepines: formulation of midazolam nasal spray. International Journal of Pharmaceutics 212, 29–40.
Lombroso, C.T. (1989) Intermittent home treatment of status and clusters of seizures. Epilepsia 30(Suppl. 2), S11–14.
Lowenstein, D.H. and Alldredge, B.K. (1998) Status epilepticus. New England Journal of Medicine 338, 970–976.
Lui, C.Y., Amidon, G.L. and Goldberg, A. (1991) Intranasal absorption of flurazepam, midazolam, and triazolam in dogs. Journal of Pharmacological Science 80, 1125–1129.
Lynch, B.A., Lambeng, N., Nocka, K., Kensel-Hammes, P., Bajjalieh, S.M., Matagne, A. and Fuks, B. (2004) The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proceedings of the National Academy of Sciences USA 101, 9861–9866.
Mahmoudian, T. and Zadeh, M.M. (2004) Comparison of intranasal midazolam with intravenous diazepam for treating acute seizures in children. Epilepsy and Behavior 5, 253–255.
Malinovsky, J.M., Lejus, C., Servin, F., Lepage, J.Y., Le Normand, Y., Testa, S., Cozian, A. and Pinaud, M. (1993) Plasma concentrations of midazolam after i.v., nasal or rectal administration in children. British Journal of Anaesthesia 70, 617–620.
Malinovsky, J.M., Populaire, C., Cozian, A., Lepage, J.Y., Lejus, C. and Pinaud, M. (1995) Premedication with midazolam in children. Effect of intranasal, rectal and oral routes on plasma midazolam concentrations. Anaesthesia 50, 351–354.
Manford, M., Fish, D.R. and Shorvon, S.D. (1996) An analysis of clinical seizure patterns and their localizing value in frontal and temporal lobe epilepsies. Brain 119(1), 17–40.
Martinez, C., Sullivan, T. and Hauser, W.A. (2009) Prevalence of acute repetitive seizures (ARS) in the United Kingdom. Epilepsy Research 87, 137–143.
Mealey, K.L. and Boothe, D.M. (1995) Bioavailability of benzodiazepines following rectal administration of diazepam in dogs. Journal of Veterinary Pharmacology and Therapeutics 18, 72–74.
Meierkord, H., Boon, P., Engelsen, B., Gocke, K., Shorvon, S., Tinuper, P. and Holtkamp, M. (2006) EFNS guideline on the management of status epilepticus. European Journal of Neurology 13, 445–450.
Milton, J.G., Gotman, J., Remillard, G.M. and Andermann, F. (1987) Timing of seizure recurrence in adult epileptic patients: a statistical analysis. Epilepsia 28, 471–478.
Mitchell, W.G. (1996) Status epilepticus and acute repetitive seizures in children, adolescents, and young adults: etiology, outcome, and treatment. Epilepsia 37(Suppl. 1), S74–80.
Mitchell, W.G., Conry, J.A., Crumrine, P.K., Kriel, R.L., Cereghino, J.J., Groves, L. and Rosenfeld, W.E. (1999) An open-label study of repeated use of diazepam rectal gel (Diastat) for episodes of acute breakthrough seizures and clusters: safety, efficacy, and tolerance. North American Diastat Group. Epilepsia 40, 1610–1617.
Modur, P.N., Milteer, W.E. and Zhang, S. (2010) Sequential intrarectal diazepam and intravenous levetiracetam in treating acute repetitive and prolonged seizures. Epilepsia 51, 1078–1082.
Monteiro, R., Adams, V., Keys, D. and Platt, S.R. (2012) Canine idiopathic epilepsy: prevalence, risk factors and outcome associated with cluster seizures and status epilepticus. Journal of Small Animal Practice 53, 526–530.
Morimoto, K., Tabata, H. and Morisaka, K. (1987) Nasal absorption of nifedipine from gel preparations in rats. Chemical and Pharmaceutical Bulletin (Tokyo) 35, 3041–3044.
Pathophysiology and Management of Cluster Seizures
Morton, L.D., Rizkallah, E. and Pellock, J.M. (1997) New drug therapy for acute seizure management. Seminars in Pediatric Neurology 4, 51–63.
Muñana, K.R., Thomas, W.B., Inzana, K.D., Nettifee-Osborne, J.A., Mclucas, K.J., Olby, N.J., Mariani, C.J. and Early, P.J. (2012) Evaluation of levetiracetam as adjunctive treatment for refractory canine epilepsy: a randomized, placebo-controlled, crossover trial. Journal of Veterinary Internal Medicine 26, 341–348.
Musulin, S.E., Mariani, C.L. and Papich, M.G. (2011) Diazepam pharmacokinetics after nasal drop and atomized nasal administration in dogs. Journal of Veterinary Pharmacology and Therapeutics 34, 17–24.
Najafabadi, A.R., Moslemi, P. and Tajerzadeh, H. (2004) Intranasal bioavailability of insulin from carbopolbased gel spray in rabbits. Drug Delivery 11, 295–300.
Nordt, S.P. and Clark, R.F. (1997) Midazolam: a review of therapeutic uses and toxicity. Journal of Emergency Medicine 15, 357–365.
O’Regan, M.E., Brown, J.K. and Clarke, M. (1996) Nasal rather than rectal benzodiazepines in the management of acute childhood seizures? Developmental Medicine and Child Neurology 38, 1037–1045.
Pakozdy, A., Leschnik, M., Sarchahi, A.A., Tichy, A.G. and Thalhammer, J.G. (2010) Clinical comparison of primary versus secondary epilepsy in 125 cats. Journal of Feline Medicine and Surgery 12, 910–916.
Papich, M.G. and Alcorn, J. (1995) Absorption of diazepam after its rectal administration in dogs. American Journal of Veterinary Research 56, 1629–1636.
Patterson, E.E., Goel, V., Cloyd, J.C., O’Brien, T.D., Fisher, J.E., Dunn, A.W. and Leppik, I.E. (2008) Intramuscular, intravenous and oral levetiracetam in dogs: safety and pharmacokinetics. Journal of Veterinary Pharmacology and Therapeutics 31, 253–258.
Payne, K., Mattheyse, F.J., Liebenberg, D. and Dawes, T. (1989) The pharmacokinetics of midazolam in paediatric patients. European Journal of Clinical Pharmacology 37, 267–272.
Perry, M. (1954) Gastrointestinal influences on seizures in the institutionalized epileptic. International Record of Medicine and General Practice Clinics 167, 489–498.
Platt, S.R., Randell, S.C., Scott, K.C., Chrisman, C.L., Hill, R.C. and Gronwall, R.R. (2000) Comparison of plasma benzodiazepine concentrations following intranasal and intravenous administration of diazepam to dogs. American Journal of Veterinary Research 61, 651–654.
Podell, M. (1995) The use of diazepam per rectum at home for the acute management of cluster seizures in dogs. Journal of Veterinary Internal Medicine 9, 68–74.
Podell, M., Smeak, D. and Lord, L.K. (1998) Diazepam used to control cluster seizures in dogs. Journal of Veterinary Internal Medicine 12, 120–121.
Poukas, V.S., Pollard, J.R. and Anderson, C.T. (2011) Rescue therapies for seizures. Current Neurology and Neuroscience Reports 11, 418–422.
Powers, D.L., Seigler, R.S. and Kilgore, D.G. (1991) The effect of diazepam enema on the rectal mucosa of rats. Pediatric Emergency Care 7, 152–153.
Prasad, K., Krishnan, P.R., Al-Roomi, K. and Sequeira, R. (2007) Anticonvulsant therapy for status epilepticus. British Journal of Clinical Pharmacology 63, 640–647.
Rey, E., Delaunay, L., Pons, G., Murat, I., Richard, M.O., Saint-Maurice, C. and Olive, G. (1991) Pharmacokinetics of midazolam in children: comparative study of intranasal and intravenous administration. European Journal of Clinical Pharmacology 41, 355–357.
Romeo, V.D., Demeireles, J., Sileno, A.P., Pimplaskar, H.K. and Behl, C.R. (1998) Effects of physicochemical properties and other factors on systemic nasal drug delivery. Advanced Drug Delivery Reviews 29, 89–116.
Rosciszewska, D., Buntner, B., Guz, I. and Zawisza, L. (1986) Ovarian hormones, anticonvulsant drugs, and seizures during the menstrual cycle in women with epilepsy. Journal of Neurology, Neurosurgery, and Psychiatry 49, 47–51.
Rose, A.B., Mccabe, P.H., Gilliam, F.G., Smith, B.J., Boggs, J.G., Ficker, D.M., Moore, J.L., Passaro, E.A. and Bazil, C.W. (2003) Occurrence of seizure clusters and status epilepticus during inpatient video-EEG monitoring. Neurology 60, 975–978.
Ruhenstroth-Bauer, G., Baumer, H., Kugler, J., Spatz, R., Sonning, W. and Filipiak, B. (1984) Epilepsy and weather: a significant correlation between the onset of epileptic seizures and specific atmospherics – a pilot study. International Journal of Biometeorology 28, 333–340.
Saito, M., Munana, K.R., Sharp, N.J. and Olby, N.J. (2001) Risk factors for development of status epilepticus in dogs with idiopathic epilepsy and effects of status epilepticus on outcome and survival time: 32 cases (1990–1996). Journal of the American Veterinary Medical Association 219, 618–623.
Sanchez Fernandez, I., Ramgopal, S., Powell, C., Gregas, M., Zarowski, M., Shah, A., Vendrame, M., Alexopoulos, A.V., Kothare, S.V. and Loddenkemper, T. (2012) Clinical evolution of seizures: distribution across time of day and sleep/wakefulness cycle. Journal of Neurology. [epub 2 Oct]
Scheepers, M., Scheepers, B., Clarke, M., Comish, S. and Ibitoye, M. (2000) Is intranasal midazolam an effective rescue medication in adolescents and adults with severe epilepsy? Seizure 9, 417–422.
Scherkl, R., Kurudi, D. and Frey, H.H. (1989) Clorazepate in dogs: tolerance to the anticonvulsant effect and signs of physical dependence. Epilepsy Research 3, 144–150.
Schwartz, M., Munana, K.R., Nettifee-Osborne, J.A., Messenger, K.M. and Papich, M.G. (2012) The pharmacokinetics of midazolam after intravenous, intramuscular, and rectal administration in healthy dogs. Journal of Veterinary Pharmacology and Therapeutics [epub 19 Dec].
Stevens, J.R., Lonsbury, B., Kodama, H. and Mills, L. (1969) Statistical characteristics of spontaneous seizure discharges recorded by radiotelemetry over 24 hour periods in man. Electroencephalography and Clinical Neurophysiology 27, 691.
Stevens, J.R., Kodama, H., Lonsbury, B. and Mills, L. (1971) Ultradian characteristics of spontaneous seizure discharges recorded by radio telemetry in man. Electroencephalography and Clinical Neurophysiology 31, 313–325.
Tauboll, E., Lundervold, A. and Gjerstad, L. (1991) Temporal distribution of seizures in epilepsy. Epilepsy Research 8, 153–165.
Todorov, A.B., Lesser, R.P., Uematsu, S.S., Yankov, Y.A. and Todorov, A.A., Jr (1994) Distribution in time of seizures during presurgical EEG monitoring. Neurology 44, 1060–1064.
Treiman, D.M., Meyers, P.D., Walton, N.Y., Collins, J.F., Colling, C., Rowan, A.J., Handforth, A., Faught, E., Calabrese, V.P., Uthman, B.M., Ramsay, R.E. and Mamdani, M.B. (1998) A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. New England Journal of Medicine 339, 792–798.
Van Hoogdalem, E., De Boer, A.G. and Breimer, D.D. (1991) Pharmacokinetics of rectal drug administration, Part I. General considerations and clinical applications of centrally acting drugs. Clinical Pharmacokinetics 21, 11–26.
Volk, H.A., Matiasek, L.A., Lujan Feliu-Pascual, A., Platt, S.R. and Chandler, K.E. (2008) The efficacy and tolerability of levetiracetam in pharmacoresistant epileptic dogs. Veterinary Journal 176, 310–319.
Wagner, S.O., Sams, R.A. and Podell, M. (1998) Chronic phenobarbital therapy reduces plasma benzodiazepine concentrations after intravenous and rectal administration of diazepam in the dog. Journal of Veterinary Pharmacology and Therapeutics 21, 335–341.
Weissl, J., Hulsmeyer, V., Brauer, C., Tipold, A., Koskinen, L.L., Kyostila, K., Lohi, H., Sauter-Louis, C., Wolf, M. and Fischer, A. (2012) Disease progression and treatment response of idiopathic epilepsy in Australian Shepherd dogs. Journal of Veterinary Internal Medicine 26, 116–125.
Wermeling, D.P. (2009) Intranasal delivery of antiepileptic medications for treatment of seizures. Neurotherapeutics 6, 352–358.
Wermeling, D.P., Record, K.A., Kelly, T.H., Archer, S.M., Clinch, T. and Rudy, A.C. (2006) Pharmacokinetics and pharmacodynamics of a new intranasal midazolam formulation in healthy volunteers. Anesthesia and Analgesia 103, 344–349, table of contents.
Wermeling, D.P., Record, K.A., Archer, S.M. and Rudy, A.C. (2009) A pharmacokinetic and pharmacodynamic study, in healthy volunteers, of a rapidly absorbed intranasal midazolam formulation. Epilepsy Research 83, 124–132.
Wilson, M.T., Macleod, S. and O’Regan, M.E. (2004) Nasal/buccal midazolam use in the community. Archives of Disease in Childhood 89, 50–51.
Yen, D.J., Chen, C., Shih, Y.H., Guo, Y.C., Liu, L.T., Yu, H.Y., Kwan, S.Y. and Yiu, C.H. (2001) Antiepileptic drug withdrawal in patients with temporal lobe epilepsy undergoing presurgical video-EEG monitoring. Epilepsia 42, 251–255.
24 Pathophysiology and Management of Status Epilepticus
Simon Platt BVM&S MRCVS Dipl. ACVIM (Neurology) Dipl. ECVN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
Status epilepticus (SE) is a medical emergency, which may be life-threatening to the patient and a challenge to the veterinarian. Continuous seizure activity necessitates prompt treatment to reduce the potential for significant neurologic morbidity. The correct management strategy involves prompt control of the seizures in addition to the treatment of the underlying aetiology, if this is known. A logical general therapeutic strategy should be devised and modified for each patient; the strategy must take into account the side-effects of aggressive pharmacologic treatment, the potential for a fatal underlying disease, the cost of therapy, the systemic effects of continuous seizure activity and the potential for irreversible cerebral damage (Fig 24.1).
Definition
Status epilepticus is defined as 5 min or more of continuous clinical and/or electroencephalographic seizure activity or recurrent seizure activity without recovery of normal physical and mental status between seizures (Knake et al., 2009; Huff and Fountain, 2011; Brophy et al., 2012; Trinka et al., 2012). SE should be classified as either convulsive SE (convulsions that are associated with rhythmic jerking of the extremities) or non-convulsive SE (seizure activity seen on EEG without the clinical findings associated with convulsive SE; Brophy et al., 2012). Refractory SE is defined as SE that does not respond to the standard treatment regimens, such as initial benzodiazepine followed by another anti-epileptic medication (AEM) (Rossetti and Lowenstein, 2011; Brophy et al., 2012).
In SE, seizures recur before full recovery from pathophysiologic alterations in brain function induced by the last seizure (Alldredge and Lowenstein, 1999; Lowenstein, 1999, 2005, 2006). If recurrent convulsions are allowed to persist without treatment or with inadequate treatment, a progressive diminution of convulsive activity may be seen such that the motor manifestations of SE become increasingly subtle. In this state, the patient may exhibit profound stupor or coma, with convulsive activity consisting of only subtle twitches of the extremities or body (Alldredge and Lowenstein, 1999; Lowenstein, 1999, 2005, 2006).
Pathophysiology of Status Epilepticus
See Chapter 1 for seizure pathophysiology.
The basic pathophysiology of SE involves a failure of mechanisms that usually stop an isolated seizure. This failure can arise from abnormal excessive excitation or ineffective
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
Brain: lctal or seizure discharges can occur without any physical manifestation.
Heart: Arrythmias can be due to hypoxic damage as well as the systemic abnormalities such as acidosis
Allison 1999UGA
Kidneys: Susceptible to poor perfusion and in combination with rhabdomyolysis can lead to acute renal failure.
Bladder: Urine production needs to be monitored for production rate and muscle breakdown products.
Skeletal muscle: Susceptible to rhabdomyolysis when the patient becomes hyperthermic.
Fig. 24.1. Status epilepticus will affect multiple body systems, which should be monitored closely after the seizure activity has stopped.
recruitment of inhibition. It is likely that numerous mechanisms are involved depending on the underlying cause (Rajasekaran et al., 2010; Huff and Fountain, 2011). Recent experimental work has suggested that the failure of inhibition may be caused by a shift in the functional properties of the GABA receptor that occurs as seizures become prolonged (Rajasekaran et al., 2010; Huff and Fountain, 2011). Repetitive neuronal firing imposes a massive metabolic demand, which is exacerbated by glutamate-mediated excitotoxicity and decreased GABA inhibitory neurotransmission (Hasegawa et al., 2004). This has become known as the excitotoxic theory of neuronal damage.
Many molecular signals are triggered by SE, activating receptors in neuronal membranes. Activation of the NMDA receptor has been shown to play a key role in neuronal signaling and delayed neuronal death (Rajasekaran et al., 2010; Huff and Fountain, 2011). It has been shown that NMDA receptors become activated during continuous neuronal stimulation, and in several animal models NMDA receptor antagonists have been shown to block or delay seizure activity (Rajasekaran et al., 2010; Huff and Fountain, 2011). However, little is known about the receptor’s precise role. Excessive concentrations of the excitatory amino-acid glutamate cause NMDA receptors to open cation channels to calcium. Large concentrations of calcium enter the neuron and then induce a cascade of intracellular neurochemical events that can kill the cells. Other possible neurotoxic substances released during SE include aspartate, free fatty acids, arachidonic acid and free radicals (Holopainen, 2008). Further to these changes potentially responsible for extensive seizure activity becoming refractory to medication, recent work has also demonstrated that a significant up-regulation of P-glycoprotein in brain tissue exists in dogs exhibiting SE (Pekcec et al., 2009).
Certain areas of the brain are more sensitive to the detrimental effects of SE. The underlying biochemical reason for this increase in sensitivity is complex and not fully understood. One theory is that there is a mechanism mediated through glutamate’s interaction with NMDA receptors. Administration of exogenous glutamate results in a similar distribution of neuronal damage as that in seizure-induced damage. Two areas sensitive to neuronal
Pathophysiology and Management of Status Epilepticus
necrosis in SE include the pyramidal cells of the hippocampus and the amygdala (Fig. 24.2). Both of these regions are rich in GABA, the major inhibitory neurotransmitter of the brain. Therefore, destruction of these regions predisposes the animal to future episodes of SE and can make long-term seizure control difficult.
Brain injury, during prolonged seizure events, also may relate to a mismatch between substrate demand and supply. Compensatory factors may be unable to meet the considerable metabolic demand placed on the brain during seizures. SE lasting longer than 30 min can cause brain damage, especially in the limbic structures. In several animal models of SE, histopathologic evidence of neuronal damage was identified within CA1 and CA3 sectors of the hippocampus, layers 3, 5, and 6 of neocortex, purkinje cells within the cerebellum, the thalamus and amygdala following prolonged seizure activity (Lado et al., 2002). Animal models of SE have also demonstrated the deleterious role that hyperthermia, hypoxia and hypotension play in creating further neuronal damage. However, observation of neuronal changes in well-ventilated animals in which adequate glucose levels have been maintained suggests that ongoing seizure activity itself substantially contributes to neuronal damage.
Causes of SE
Dogs
The overall prevalence of SE in veterinary medicine has not been evaluated, however the prevalence of dogs with either SE or cluster seizures has been estimated to be 0.44% of the total hospital admissions. Approximately 59% of dogs with epilepsy of any cause will have one or more episodes of SE during their lifetime (Saito et al., 2001).
Any type of cerebral disease can potentially cause SE and indeed continuous seizure activity may be the first sign of an underlying disease process in the brain. The differentials for SE include idiopathic epilepsy (IE) and so the presentation of a patient in a continuous seizure state does not infer any information about the predisposing aetiology. Approximately one in eight humans with IE will develop SE at some point in their lives with an overall prevalence of 20/100,000 Caucasians in industrialized countries (Knake et al., 2009); approximately 5% of dogs with IE will develop SE at some point in their lives (Monteiro et al., 2012). Certain breeds with IE, such as Australian shepherd dogs and Border collies, may be more predisposed to exhibiting SE than others for which there may be a genetic basis (Hulsmeyer et al., 2010; Weissl et al., 2012).
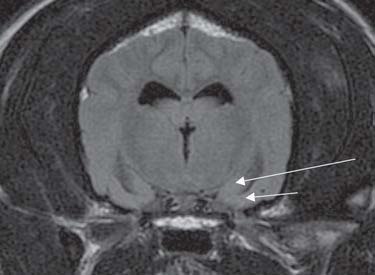
Several veterinary studies have investigated the epidemiology of SE in dogs (Bateman and Parent, 1999; Saito et al., 2001; Platt and Haag, 2002). A specific cause for the seizures cannot be determined in 25–30% of the cases (Bateman and Parent, 1999; Saito et al., 2001; Platt and Haag, 2002). Approximately 27% of the cases are diagnosed with IE (Platt and Haag, 2002). Secondary (structural) epilepsy, such as inflammatory disease or neoplasia, is identified as the cause of the seizures in 32–35% of the cases (Bateman and Parent, 1999; Saito et al., 2001; Platt and Haag, 2002; Pakozdy et al., 2008, 2010; Zimmermann et al., 2009a); cerebrospinal fluid (CSF) abnormalities have been documented in 36–74% of cases and neuroimaging examination (magnetic resonance imaging or computed tomography) abnormalities have been documented in 46–76% of cases, but both diagnostics may be abnormal due to protracted seizure activity (see Chapter 10) (Bateman and Parent, 1999; Saito et al., 2001; Platt and Haag, 2002). Reactive seizures (RS) are responsible in approximately 7–11% of the cases (Zimmermann et al., 2009a; Brauer et al., 2011). Dogs with RS are considered to have normal brain structure, but the seizures are caused by intoxication or metabolic abnormalities. Intoxications with compounds such as metaldehyde, strychnine, moxidectin and theobromine can result in seizure activity and such toxins should be suspected when SE is seen in a dog without prior history of seizure activity (Yas-Natan et al., 2007; Crandell and Weinberg, 2009; Zimmermann et al., 2009b; Jull et al., 2011).
Chronic processes that cause SE in dogs include pre-existing epilepsy in which SE is caused by breakthrough seizures or the dis-continuation of AEMs. A low AEM concentration has been determined to be the cause of the SE in approximately 6% of presented canine cases (Bateman and Parent, 1999).
Cats
Limited work has been done evaluating the causes of SE in cats. One study evaluating 125 cats with primary and secondary causes of seizure activity documented SE in 19% and 40% of cases, respectively (Pakozdy et al., 2010). Specific causes of SE were not discussed in this study. An earlier study investigated 88 cats with seizure disorders and documented 19% with generalized SE (Schriefl et al., 2008). Of the 17 cats described in this publication, ten had structural epilepsy (22% of all cats with symptomatic disease), six had RS (30% of those with reactive seizures) and one had idiopathic seizures (4% of all cats with idiopathic disease) (Schriefl et al., 2008).
Outcome of SE
An unbiased mortality rate of dogs or cats with SE is unknown, because many animals are euthanized prior to aggressive diagnosis and treatment, although it has been estimated to be approximately 25% (Bateman and Parent, 1999). The overall mortality rate among human adults with SE varies in the literature from 3 to 22% (Claassen et al., 2002b; Rossetti et al., 2006; Novy and Rossetti, 2010). This is difficult to interpret given the variety of underlying problems that can cause SE. However, patient age at diagnosis, aetiology of SE, severity of the underlying disease and duration of SE are the main short-term predictors of increased mortality (Wu et al., 2002; Garzon et al., 2003; Trinka et al., 2012). Marked functional impairment following status can be seen in as high as 39% of human patients 3 months after the event (Legriel et al., 2010).
The prognosis of dogs with SE due to intoxication can be very good (approximately 85% survival) and when followed long term, these dogs do not tend to exhibit further seizure activity (Zimmermann et al., 2009b, 2010; Brauer et al., 2011; Jull et al., 2011). No significant associations have been observed between the outcome of dogs with SE and the breed of the dog, the dog’s age at onset of seizure activity, or the type of seizure activity at admission. Dogs with IE that exhibited SE did have a worse prognosis than those juvenile dogs with IE that did not exhibit SE (Arrol et al., 2012).
A poor prognosis exists when the SE in dogs is due to an underlying cerebral neoplasia or severe inflammatory disease such as granulomatous meningoencephalitis. The prognosis also becomes poor when the SE cannot be controlled within 6 h of its onset. In addition to the above correlations, hospital visits during which focal motor SE is documented have a significant association with a poor outcome for dogs.
The mean duration of hospitalization for dogs with SE or cluster seizures is 51.6 ± 42.6 h, which can depend on the underlying disease and has serious financial implications in many cases; it also needs to emphasized at this point that recurrent SE is a likelihood in many patients after initial stabilization and hospitalization.
A study of 88 cats which identified 19% with generalized SE also attempted to evaluate a possible effect of SE on survival time (Schriefl et al., 2008). Results of a log-rank test that compared 1-year survival rates of cats with SE and cats without SE indicated that cats in which SE occurred had a significantly (P = 0.04) shorter survival time and SE occurred more commonly in cats with symptomatic or
Pathophysiology and Management of Status Epilepticus
reactive seizures, compared with cats with idiopathic seizures.
Clinical Features of SE
The mean age of dogs presented in SE has been estimated to be 4.2–5.0 years (range 0.15–15.0 years) (Bateman and Parent, 1999; Saito et al., 2001; Platt and Haag, 2002). Results of one study indicate that there is a male sex predilection for primary epilepsy but no statistical gender prevalence has been documented in dogs with SE. There have been no significant breed associations documented with this condition, apart from one study which concluded that the English foxhound, pug, teacup poodle, Boston terrier and Lakeland terrier were over-represented (Bateman and Parent, 1999).
Patients with SE usually have clinically obvious seizures, such as tonic, clonic, or tonicclonic movements of the extremities. This is generalized seizure activity and is usually accompanied by a marked impairment of consciousness. Typically, there is gradual recovery of consciousness following each convulsion, but if the patient has not recovered fully to baseline before the next convulsion occurs, the patient is considered to be in generalized tonic-clonic SE. Non-convulsive focal SE is well recognized in humans; although variations of this do exist in dogs and cats, this type of SE has not been well documented clinically or electroencephalographically in veterinary medicine.
Systemic Features of SE
Several systemic changes occur during SE including hypertension, tachycardia, hypoglycaemia, acidosis and hyperthermia (Rossetti and Lowenstein, 2011). Some of these changes, if prolonged, can be life-threatening and undoubtedly contribute to the morbidity and mortality of patients with SE. Early recognition, appropriate intervention and prevention of such complications are imperative during the treatment of SE.
The initial physiologic response is a massive release of catecholamines into the circulation, which results in increased systemic, pulmonary and left atrial blood pressure, central venous pressure, heart rate, plasma glucose concentration and an increase in cardiac arrhythmias. There is also an increase in cerebral blood flow (CBF) in the range of 200% to 700%, presumably compensatory for the increased metabolic demands of the brain. As the seizure persists, however, systemic blood pressure tends to decrease often to hypotensive levels; CBF is also reduced, although it remains higher than normal levels. As seizure activity continues, the brain’s metabolic rate remains high, but the observed increase of CBF is incapable of supplying adequate substrate and oxygen to meet these metabolic demands. This is compounded by the fact that intracranial pressure increases early in SE and remains elevated throughout prolonged seizures; cerebral herniation has even been observed as a consequence of these processes in animal studies.
Respiratory function is frequently impaired in early SE due to impaired ventilation, autonomic dysfunction, excessive salivation and tracheobronchial secretions; the end result is commonly hypoxia. Hypoxia is responsible for most of the complications seen in SE resulting in cerebral lactic acidosis, impaired cardiac ventricular function, reduced cardiac output, and hypotension, which combine to compromise neuronal and tissue cell function. Systemic metabolic and respiratory acidosis are also commonly found with SE.
In patients with uncontrolled seizures, generalized muscular contractions are responsible for an increase in body temperature and even hyperthermia, which can lead to rhabdomyolysis and myoglobinuria. In combination with hypotension and severe metabolic acidosis, myoglobinuria may compromise renal function, resulting in acute renal failure (Fig. 24.1).
Management of SE
Status epilepticus is a danger to the patient and a treatment challenge for the clinician. Although no evidence exists to indicate that early initiation of appropriate treatment improves the outcome in dogs and cats with seizures, such evidence does exist for human medicine and remains an important basic tenet of treatment (Knake et al., 2009). The main goals of the treatment of SE are to simultaneously:
(iii) terminate the seizure activity safely and quickly while minimizing treatment-related morbidity (Lowenstein, 1999, 2006; Lowenstein and Cloyd, 2007; Rossetti, 2010; Rossetti and Lowenstein, 2011; Brophy et al., 2012).
A team approach to patients in SE will be beneficial to accomplishing emergency stabilization, therapeutic intervention and diagnostic investigation simultaneously. Concurrently, a medical history should be obtained from the owners or retrieved from available medical records.
Identification and treatment of underlying causal factors
The precipitating factors in a case of SE must always be vigorously sought and treated to facilitate seizure control and to be certain that the underlying cause is treated before it results in irreversible cerebral damage (Box 24.1).
Standard laboratory blood tests should be performed including evaluations for glucose, sodium and calcium level abnormalities, renal and hepatic dysfunction, as well as AEM serum levels and a toxicity screen if deemed appropriate. It should be noted that liver enzymes may be increased shortly after seizure activity because of the effects of hypoxia and hypotension.
If hypoglycaemia (i.e. glucose concentration <60 mg/dl; <3 mmol/l) is a potential cause of SE or if blood glucose determination is unavailable, 500 mg/kg body weight of 50% dextrose (1 ml/kg), preferably diluted to 25%, should be administered intravenously over 15 min. It should be remembered that hyperglycaemia can be detrimental to the brain in the hypoxic environment created by SE. To counteract this potential, administration of thiamine (vitamin B1) at 25–50 mg per animal intramuscularly, should precede parenteral glucose administration. Thiamine is essential as a coenzyme in glucose utilization by the brain. If intravenous glucose therapy is difficult to perform, oral administration of a sugar solution can be a useful substitute.
If encephalitis is suspected, a cerebrospinal fluid analysis should be considered as soon after seizure stabilization occurs. Patients with new-onset seizures should be considered for brain imaging procedures such as computed tomographic (CT) scanning or a magnetic resonance imaging (MRI) procedure. However, a normal CT scan does not exclude the possibility of cerebral pathology. Lesions in the piriform/temporal lobe region of the brains of
Box 24.1. Diagnostic investigation of status epilepticus patient.
• Dynamic bile acids assessment
Pathophysiology and Management of Status Epilepticus
dogs which have experienced SE have been described (see Chapter 10). All of the lesions have varying hyperintensity on T2-weighted images and hypointensity on T1-weighted images, and all of them partially or completely resolve after variable periods of time (from 10 days to 18 weeks) without seizure activity (Mellema et al., 1999). The increased signal intensity on T2-weighted images represents an increase in relative water content of the brain tissue. Cytotoxic and vasogenic oedema may both be responsible for this signal intensity seen following seizure activity and initially may only be seen on diffusion weighted imaging (Hasegawa et al., 2003).
Maintain vital functions
Treatment may have to begin even before the diagnostic evaluation in practice situations where help is limited. Medical and neurologic examinations should be performed concomitant with the management of the patient.
The initial care of a patient with SE involves basic medical emergency measures, namely the ABCs of life support (Box 24.2).
Oxygenation, airway and patient acid-base status
Hypoxia must be corrected in order for recovery to occur. Airway management and respiratory monitoring in the SE patient can be difficult prior to termination of the seizure event. If possible, the airway patency should be maintained while the patient remains unresponsive. The administration of 100% oxygen by the use of a non-rebreathing mask or an intranasal delivery system is advised.
Arterial blood gas (ABG) monitoring is extremely useful in SE patients as marked metabolic acidosis can be prevalent in these patients during the convulsive episodes. Respiratory acidosis or hypoxia detected on the ABG should be treated immediately, whereas metabolic acidosis may resolve once the physical manifestations of the convulsions subside. Maintenance fluid therapy should be sufficient
Box 24.2. Monitoring the status epilepticus patient.
in most patients; intravenous bicarbonate therapy is rarely necessary.
Intravenous access
A large intravenous catheter should be inserted for fluid and medication administration. A maintenance rate administration of intravenous isotonic saline, with potassium chloride supplementation, may be initiated at this point. Other secondary metabolic complications such as electrolyte imbalance must be corrected.
Temperature regulation
Hyperthermia can occur frequently during SE, and even in human cases represents a manifestation of the seizures rather than evidence of an infection. Prolonged hyperthermia can be life threatening and should be treated promptly with passive cooling, especially if the temperature exceeds 40°C (104°F). In certain patients, rectal monitoring of temperature may be necessary, particularly if cooling of the patient is performed. The passive cooling should be stopped at 102°F to prevent rebound hypothermia.
Termination of the seizure activity
Although the definition of SE incorporates a 30-min duration, which is useful for research purposes, it can be misleading in terms of treatment decisions. Patients with seizures lasting more than 10 min should be treated. Most self-limiting generalized convulsions stop within 3 min, and almost all stop by 5 min from onset.
Several AEMs are effective in the treatment of SE. The timing, time of onset, route and adequacy of dosages of the medications used are probably more important than the specific choice of drugs in determining success of therapy. Although the use of a single AEM of sufficient dosage is always preferred, often more than one medication is needed to achieve all of the therapeutic goals. Familiarity with the various AEMs used and their possible side-effects, including cardiovascular and respiratory depression, is necessary in the treatment of SE.
Principles of AEM Treatment
Factors determining the AEM to be used in SE are its ease of administration, time of onset, duration of effect and its minimal effects on the cardio-respiratory function and level of consciousness. Unfortunately no single AEM is ideal. The rate of brain entry of a medication is directly proportional to nonprotein-bound drug serum concentration, lipid solubility and cerebral blood flow. Therefore SE is treated by intravenous (IV) infusion (to obtain high serum concentration) of lipid-soluble AEMs. When a medication is selected to be used, sufficient time must be allowed for the AEM to act before more of the same medication is used. If a single agent does not control the seizures, a second AEM may be needed. If the combined effects of two or three AEMs do not achieve cessation of seizures, alternative aggressive treatment such as general anaesthesia should be considered (Fig. 24.3).
The goals of anti-epileptic therapy in SE are to achieve cessation of clinical and electrical seizure activity and prevent its recurrence. Intravenous AEM treatment for SE should be started without delay. This is necessary based upon the relationship between duration of SE and the extent of neurologic morbidity. This approach is also based upon experimental animal models that suggest that SE becomes progressively less responsive to treatment with diazepam (Rossetti and Lowenstein, 2011).
Benzodiazepines
Benzodiazepines (diazepam, lorazepam, midazolam and clonazepam) are potent, fast-acting AEMs and therefore (particularly diazepam) are often the preferred initial therapy in SE (Brophy et al., 2012). When skilled technicians, veterinary nurses or veterinarians are present, intravenous (IV) administration is preferred (Lowenstein and Alldredge, 1998; Lowenstein and Cloyd, 2007). However, benzodiazepines can be administered via intramuscular (IM), rectal, nasal or buccal routes when IV therapy is not feasible.
Nasal and buccal midazolam has become a promising first-line treatment option for
Pathophysiology and Management of Status Epilepticus

SEIZURES STOPPED?
NO
REPEAT × 2–3 OR DIAZEPAM INFUSION (0.1–0.5 mg/kg IV diluted in
dextrose saline given as a maintenance fluid requirement)
• Continue phenobarbital, levetiracetam or bromide load
YES
• Propofol 1–2 mg/kg IV bolus or 0.1–0.6 mg/kg/min titrated to effect (up to 6 mg/kg/h as a CRI) or
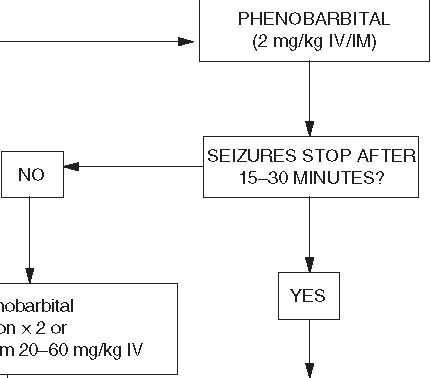
![]() • Continue phenobarbital bolusing
• Continue phenobarbital bolusing
up to a maximum of 24 mg/kg in followed by 5 mg/kg/h CRI or
24 h IV inclusive of daily
maintenance dose where
applicable
Fig. 24.3. Algorithm for the anti-epileptic treatment in dogs and cats with status epilepticus.
out-of-hospital treatment of human SE (Wallace, 1997; Mahmoudian and Zadeh, 2004). Rapid uptake and high bioavailability were demonstrated in a healthy canine study comparing the pharmacokinetics of intravenous and nasal midazolam (Eagleson et al., 2012). A recent study of human SE found that IM administration of midazolam was preferable to IV therapy of lorazepam, which is often the preferred AEM for SE (Silbergleit et al., 2012).
The recommendation for veterinary SE from the author is to immediately consider IM midazolam where IV access is difficult to immediately achieve, as is so often the case.
None of the benzodiazepines are effective for chronic control of SE. They are a widely used class of sedative/tranquilizer and anxiolytic agents that differ widely in their time course and their central effects. These differences may be related to their pharmacokinetics, especially their distribution into and out of the central nervous system, which has been related to the drugs’ lipophilicity and plasma protein binding. The lipophilicity of these compounds determines their rapid brain penetration after IV administration. Although the penetration is rapid, distribution equilibrium among all regions takes longer.
Their primary pharmacologic actions are probably related to a benzodiazepine-receptormediated enhancement of g-aminobutyric acid (GABA)-ergic transmission, both pre- and post-synaptically. Benzodiazepines do not seem to alter the synthesis, release or metabolism of GABA but rather potentiate its action at the receptor. The resultant augmented flow of chloride ions into cells decreases the ability of the cell to initiate an action potential. It seems that benzodiazepines prevent the spread of seizure rather than suppress the focus.
Adverse effects of IV benzodiazepines include respiratory depression, hypotension and impaired consciousness. However, it has been suggested that there is a low incidence of respiratory depression with benzodiazepines because of the low density of binding sites in the brainstem.
Specific pharmacologic details about each of the benzodiazepines can be found in Chapter 21.
Phenytoin sodium (Diphenylhydantoin)
Although this AEM is used in SE in humans, its IV injection can cause severe hypotension in dogs and its IM absorption is extremely variable (Smith and Lomas, 1978; Frey and Loscher, 1985). Intramuscular injection of phenytoin can also cause adverse local reactions caused by precipitation at the site, including soft-tissue sloughing. The half-life of phenytoin in the dog is variable, being as short as 3.65 h after an 11 mg/kg IV dose (Frey and Loscher, 1985; Overduin et al., 1989). In the cat, phenytoin can have an effective duration of up to 108 h, and this may be related to a decreased ability to conjugate the compound to glucuronic acid (Hassell et al., 1984).
Fosphenytoin (FOS)
Fosphenytoin sodium is a phosphate ester pro-drug of phenytoin developed as a refinement of parenteral phenytoin. After administration, phenytoin is cleaved from the prodrug by phosphatases in the blood stream and several other organs. However, fosphenytoin is freely soluble in aqueous solutions and rapidly absorbed by the IM route. Fosphenytoin has no known anti-epileptic action before conversion. At present, there are no veterinary studies using this medication clinically as AEM but a trial is underway and initial canine pharmacokinetic data has been published (Leppik et al., 2011). An initial test dose of 25 mg/kg produced unbound phenytoin concentrations in the 3.0–4.5 mg/ml range 30 min following dosing (4.6–6.3 mg/ml 15 min following dosing) (Leppik et al., 2011). These unbound levels were higher than expected and were associated with ataxia and, in one dog, vomiting. A dose of 15 mg/kg was tolerated, and because of lower protein binding in dogs, produced unbound phenytoin concentrations (2.0–2.5 mg/ml) that are in the range observed when treating humans with SE even though the total levels were in the 12–13 mg/ml range, lower than in humans following 18 mg/kg. Approximately 80% of FOS was protein-bound 15 min following dosing, compared to ~90– 95%, which has been reported for humans (Leppik et al., 2011). Furthermore, ~80% of fosphenytoin was converted to phenytoin by 30 min, suggesting rapid metabolism similar to that observed in humans.
Barbiturates
Phenobarbital
Phenobarbital (PB) is a safe, inexpensive AEM that may be administered orally, intravenously or intramuscularly (see Chapter 13). PB increases the seizure threshold required for seizure discharge and acts to decrease the spread of the discharge to neighbouring neurons (Thomas, 2010). The actions of this AEM are primarily due to the enhancement of the inhibitory effects of GABA post-synaptically.
Pathophysiology and Management of Status Epilepticus
There is also some inhibition of both glutamate activity and the flux of calcium across the neuronal membranes.
The distribution of PB to the central nervous system may take up to 30 min, because of weaker lipophilicity in comparison with diazepam. This will need to be considered if the animal is still exhibiting generalized seizure activity. The recommended loading dose in dogs is 12 to 24 mg/kg IV if immediate therapeutic concentrations are desired, but this can induce a profound stupor with concurrent suppression of the cardiovascular and respiratory system. Alternatively, the dose can initially be 2 mg/kg IV, repeating the dose every 20–30 min to effect and to a maximum total 24-h dose of 24 mg/kg. The parenteral form can also be given IM, which is recommended if diazepam has already been administered. This will avoid the potentiation of profound respiratory and cardiovascular depression. The loading dose in cats for this drug is not as clear but it is suggested to be similar, with caution being taken to avoid repeat boluses if sedation or hyperexcitability is noted. The prior use of diazepam in cats may make excessive sedation more likely, which is why the use of levetiracetam is preferred by some clinicians for SE treatment in cats (see below and Chapter 16). This is also the case in dogs or cats that have a pre-existing liver disease or exhibit seizure activity after porto-systemic shunt surgery.
The depressant effects of PB on respiratory drive, level of consciousness and blood pressure may complicate management of the SE patient, especially when administered after benzodiazepines. For these reasons, tracheal intubation may be necessary during IV administration of PB.
Treatment of Refractory SE
Status epilepticus that does not respond to a benzodiazepine or PB is considered refractory and requires more aggressive treatment (Claassen et al., 2002a; Bleck, 2005; Abend and Dlugos, 2008). Refractory SE (RSE) occurs in up to 44% of all human patients with SE and mortality can be up to 39%, which is two to three times higher than non-refractory SE (Knake et al., 2009; Novy et al., 2010; Rossetti and Lowenstein, 2011). Potential reasons for resistant seizure activity include inadequate anticonvulsant doses, an uncorrected metabolic abnormality or the presence of an intracranial disease, such as a tumour. These patients often represent a difficult therapeutic problem and success depends on rapid intervention (Rossetti and Lowenstein, 2011). Short-acting anaesthetic drugs are the most commonly used agents for treating resistant SE, as they have a rapid onset of action, short half-lives and cause reductions in cerebral metabolic rates. These drugs should be used only in an intensive-care setting because of the need for continuous blood pressure monitoring and, ideally, central venous pressure monitoring. General anaesthesia prevents tonic-clonic movements and allows control of respiration. More recent human work has proposed the use of combination therapies of the newer antiseizure medications for RSE in an attempt to avoid induction of a coma. A small study of 23 humans with RSE due to brain tumours claimed relative success using a combination of phenytoin, levetiracetam and oral pregabalin (Swisher et al., 2012). No such prospective studies have been performed as yet in animals, but if effective, similar ‘up-front’ combination therapy may be worth considering in dogs or cats.
Continuous benzodiazepine infusion
This has been shown to be an effective mode of therapy in RSE in human and veterinary patients. The dose should be calculated hourly (diazepam 0.1 to 0.5 mg/kg of body weight, q1h) and is usually diluted in 0.9% saline or in 5% dextrose in water (D5W), with the volume used being equal to the maintenance fluid requirement over the hour. The dose can be delivered with an infusion pump. The dosage rate should be reduced by 50% every 6 h for at least two reductions before discontinuing the drug. Midazolam is completely water-soluble and has been shown to be an effective and safe therapy when administered by constant rate infusion. Furthermore, recent work has established that the half-life of this medication in people actually increases after prolonged administration (Rossetti and Lowenstein, 2011). No similar work has been done in veterinary medicine but it is likely the same phenomenon may exist.
Levetiracetam
Levetiracetam (20–60 mg/kg IV) is a newer AEM, which has a half-life of 3–4 h in dogs and in cats (see Chapter 16). Although it can be reserved for RSE treatment, many clinicians are becoming more comfortable with its use as a primary medication for this disorder. This is particularly true in cases that have a history of liver disease or phenobarbital toxicity.
Its IV use may be effective for 8 h at which time it can be repeated. While the binding site of the AEM, a site on a synaptic vesicle protein in neurons, has been identified, the exact mechanism of action is unknown. It is thought to act by modifying calcium-dependent exocytosis of neurotransmitters and may therefore be synergistic with phenobarbital or potentially effective where phenobarbital has not been. When used with phenobarbital, a dose at the upper end of the range of levetiracetam may be necessary. It causes minimal sedation making it desirable in treating the more refractory SE patients that already have an altered consciousness. It is not metabolized in the liver and so represents a more suitable option than phenobarbital for dogs and cats with porto-systemic shunts or liver disease. Excretion is purely renal and thus there are minimal interactions with other anticonvulsant medications; however, caution should be used in patients with deficient renal function. Levetiracetam may also have neuroprotective effects, reducing seizure-related brain damage. As for phenobarbital, the oral maintenance use of levetiracetam should follow its parenteral use once SE has been controlled.
Several human case series have demonstrated a 50–60% efficacy of IV levetiracetam as a third-line drug for SE (Aiguabella et al., 2011). A randomized, placebo-controlled, double-masked study including 19 dogs with SE or cluster seizures has shown that administration of intravenous LEV (30 or 60 mg/kg) in addition to diazepam resulted in a significantly higher responder rate (56% after LEV) compared to placebo and diazepam (10%). In addition, dogs in the placebo group required significantly more boluses of diazepam compared with the LEV group (Hardy et al., 2012). Further studies are required to investigate LEV efficacy in SE in a larger number of dogs and to determine the maximum safe dosage of IV LEV, efficacy of single versus multiple doses of LEV, the optimal dosing interval of LEV, and timing of LEV treatment relative to administration of benzodiazepines or other AEMs.
A recent human study evaluated levetiracetam against benzodiazepine administration for convulsive SE therapy and demonstrated that the drugs were equivalent in their effect; the levetiracetam group was less likely to require ventilation with a trend toward less hypotension and respiratory depression as well (Misra et al., 2012).
Propofol
In human cases of refractory SE, the use of IV infusions of anaesthetic doses of propofol, 2,6-diisopropylphenol, has become standard (Brophy et al., 2012). This approach has recently been evaluated in veterinary patients. Propofol has barbiturate- and benzodiazepinelike effects on the GABAA and NMDA receptors and can suppress CNS metabolic activity (Marik, 2004). Propofol can be administered by IV bolus (1–2 mg/kg) or by constant rate infusion (0.1–0.6 mg/kg/min titrated to effect or up to 6 mg/kg/h). The advantages of this drug over the barbiturates are its rapid clearance, chiefly eliminated by hepatic conjugation to inactive metabolites, and less profound hypotensive effects. However, this drug should be used with caution, preferably in settings where definitive airway control and haemodynamic support is possible, as hypoxaemia secondary to apnoea is a primary side-effect as is myocardial depression. Experimental animal models have documented propofol’s anti-epileptic effects.
In human medicine, a propofol infusion syndrome has been reported when propofol
Pathophysiology and Management of Status Epilepticus
has been used at high doses (>4 mg/kg/h) or for prolonged periods (>48 h). This syndrome is due to impairment of mitochondrial activity and use of free fatty acids, with resulting mismatch between energy needs and use (Rossetti and Lowenstein, 2011). Signs of this syndrome include metabolic acidosis, rhabdomyolysis, hyperkalaemia, lipaemia, renal failure, hepatomegaly and cardiovascular collapse (Vasile et al., 2003). While this syndrome has not been reported in veterinary patients the possibility exists, especially in those patients maintained on a constant rate infusion (CRI) long term. It is important to note that propofol is a phenol and thus capable of causing oxidative injury to red blood cells (RBC) of the cat resulting in Heinz body formation and haemolytic anaemia.
Ketamine
Ketamine is an N-methyl-D-aspartate (NMDA) receptor antagonist. NMDA receptor antagonists, like ketamine, are able to end the maintenance phase of chronic SE, sometimes called self-sustaining SE (Mazarati et al., 1998; Mazarati and Wasterlain, 1999). NMDA receptor activation only occurs in later phases of SE, perpetuating the seizure activity, so NMDA antagonists are suspected to be beneficial during prolonged or refractory SE (Rossetti and Lowenstein, 2011). Ketamine also may have neuroprotective effects by inhibiting NMDA receptor-mediated excitotoxicity associated with prolonged seizure activity; however, there is also some evidence that excessive antagonism of the NMDA receptors can be detrimental. Although the use of ketamine has been documented in a dog with SE (5 mg/kg IV bolus followed by 5 mg/kg/h CRI) (Serrano et al., 2006), there are currently no clinical studies documenting the effectiveness or safety of ketamine CRIs in treating veterinary patients.
Barbiturates
Thiopentone and pentobarbitone have potential, though unproven, cerebral protective effects in the management of SE. In adequate doses these drugs will almost always control the physical manifestations of seizures, but severe hypotension limits their safety. Pentobarbitone sodium, used at standard safe doses, is a general anaesthetic with negligible anticonvulsant properties. Thiopentone has been associated with a higher degree of cardiac toxicity than pentobarbitone. Pentobarbitone should be given to effect and not as a specific dose (3–15 mg/kg body weight IV), as there is tremendous individual variation in response. Patients treated with ‘barbiturate coma’ commonly require an extended period of mechanical ventilation in an intensive care setting. In general, the side effects of barbiturate coma include depression of myocardial metabolism, vasodilatation with a decrease in venous return and decreased cardiac perfusion. These effects can be minimized by the use of saline infusion and small doses of dopamine. Patients can develop poikilothermia and decreased urinary output during myocardial depression and hypotension. Neurologic evaluation is difficult because spontaneous respiratory responses and spontaneous movements cease.
Inhalational anaesthesia
Inhalational anaesthetics have been recommended as a last resort in cases of resistant SE (Shorvon and Ferlisi, 2012). The equipment and personnel necessary to administer inhalational anaesthesia may not be readily available and can be cumbersome. Isoflurane, an inhalational general anaesthetic agent, may be efficacious in the treatment of resistant SE (Mirsattari et al., 2004). Not all of the volatile anaesthetic agents have anti-epileptic potential however; enflurane may actually increase seizure activity. Isoflurane does not undergo hepatic metabolism, has a rapid onset of action and has been extensively studied. Obviously, isoflurane therapy necessitates ventilation and intensive-care monitoring, and hypotension may occur during therapy.
Lacosamide
Lacosamide (LCM) (see Chapter 20), a recently approved anticonvulsant in humans, is
available as an intravenous formulation. It has a unique profile when studied across a range of animal models of epilepsy, demonstrating an anti-epileptic effect similar to many of the newer AEMs. It is effective in the maximal electroshock model and elevates the seizure threshold in the pentylenetratazol seizure test (Stohr et al., 2007). A kindling seizure model has been used to evaluate whether LCM affects kindling-induced epileptogenesis. The rats were treated with either vehicle or different doses of LCM (3, 10, or 30 mg/kg/day) over 22–23 days during amygdala kindling. Daily administration of LCM during kindling acquisition produced a dose-dependent effect on kindling development. Although the medication was inactive at 3 mg/kg/day, significant retardation of kindling was observed at 10 mg/kg/day, at which the average number of stimulations to reach kindling criteria was increased by >90%. These data demonstrate that LCM, in addition to exerting anti-epileptic activity, has the potential to retard kindling-induced epileptogenesis (Brandt et al., 2006).
LCM also decreases self-sustaining status epilepticus (SSSE) in rats, inhibits NMDA-induced seizures in mice and is able to completely block 4-aminopyridine-induced seizures in vitro (Lees et al., 2006; Stohr et al., 2007). Spike frequency cumulative time spent in seizures significantly decreased; only separate spikes were recorded for 12 h after induction of SSSE (Wasterlain et al., 2011). In the control group, three of the six animals died, whereas, in the treatment group, all survived. Histological examination of brain sections (dorsal hippo-campus) collected 72 h after SE revealed significantly less damage in LCM-treated rats compared with control animals, suggesting LCM may be neuroprotective (Beyreuther et al., 2007).
Although RSE is known to be more resistant to treatment, with a worse prognosis with each unsuccessful attempt to treat, 64.7% of RSE human patients responded when IV lacosamide was used as an add-on therapy, with no serious side effects noted (Miro et al., 2012). LCM should be considered a future treatment option for SE but as yet clinical veterinary trials are lacking to prove its safety and efficacy.
Transition to Maintenance Therapy
Once seizure activity has been controlled and systemic stabilization has been ensured, a maintenance AEM will need to be considered. In a naïve patient phenobarbital may be used as the sole medication. In a patient with a history of SE it is recommended that the patient be loaded so that steady-state serum levels are reached as quickly as possible (see Chapter 13). If the animal was on phenobarbital prior to the episode of SE, two options exist. If the animal’s serum phenobarbital level is low, an increase in dose may be indicated. If the serum level is well within the reference range, is approaching toxic levels, or if the patient is displaying adverse effects, an additional AEM may be added. Add-on AEMs include potassium bromide, zonisamide, levetiracetam, gabapentin and pregabalin administered at standard doses. Some of the oral maintenance drugs have been evaluated for their efficacy in the treatment of SE in humans when administered using a nasogastric tube. Most recently, pregabalin was evaluated in people with non-convulsive SE (Swisher et al., 2013).
The success of the maintenance regimen is predicated by many clinical features, including the cause of SE, concurrent systemic disease and drug-drug interaction profiles.
There are no data to guide transition from continuous infusion treatment to intermittent maintenance therapy following resolution of RSE. In general, maintenance AEMs are given in doses sufficient to maintain therapeutic concentrations during and after weaning off the continuous infusion. Therapeutic concentrations may exceed published target concentrations for many AEMs and dosing should be individualized to achieve seizure control and minimize adverse effects. Patients exposed to prolonged infusions will need to be weaned down slowly due to a risk of withdrawal seizures.
Continuous EEG Monitoring in SE
The treatment of SE in the intensive care unit (ICU) in humans usually requires continuous EEG monitoring to direct treatment (Brophy et al., 2012). This diagnostic modality
Pathophysiology and Management of Status Epilepticus
is especially useful in cases of non-convulsive SE. In patients being treated with continuous infusion AEMs, in which most or all convulsive activity resolves, continuous EEG is the only way to know if treatment is successful. The use of video monitoring in conjunction with EEG in the ICU may aid EEG interpretation and help assess the presence of clinical behaviours accompanying the ictal EEG. However, no studies have been performed to formally assess efficacy of adding video to EEG in the setting of SE in veterinary or human medicine. The timing, duration and essential technical elements for EEG are also very important considerations in patients with SE. In humans, the cumulative duration of SE affects mortality and neurologic outcome (Brophy et al., 2012). Knowing this, EEG should be initiated where possible within 1 h of suspected SE in all patients. The duration of EEG monitoring if used should be 24 h after cessation of electrographic seizures or during the AEM weaning based on human trials (Claassen et al., 2002b, 2004; Vespa et al., 2007; Abend et al., 2010). EEG electrodes should be placed to sample major regions of the brain, and CT/MRI compatible electrodes may be considered (Jordan, 1995; Vespa et al., 1999; Vulliemoz et al., 2009). Due to the complexity of EEG recordings in patients with SE, the person reading the EEG should have specialized training in EEG interpretation, including the ability to analyse raw EEG as well as quantitative EEG tracings (Hirsch, 2004). However, continuous, real-time reading of EEG, especially after hours, is not available in many veterinary hospitals and institutions. Additionally, even in human medicine, EEG endpoints are controversial and so removal of seizure discharges for 24 h is all that can be advised for veterinary medicine. A recent report on the use of EEG in monitoring SE in dogs and cats described it as a useful technique allowing detection of seizures without their clinical appearance (Raith et al., 2010). For further information on the utility of EEG for seizure diagnosis and management, see Chapter 11.
Summary
Status epilepticus is a serious medical emergency that requires prompt and appropriate intervention. Maintenance of adequate vital functions with attention to airway, breathing and circulation, prevention of systemic complications and rapid termination of seizures must be coupled with investigating any underlying cause. In most patients with SE, the use of adequate dosages of first-line anti-epileptic agents allows for the successful and rapid termination of SE and avoidance of potential neurologic complications. Refractory SE requires more aggressive treatment and intense monitoring, but no single logical approach is always effective.
References
Abend, N.S. and Dlugos, D.J. (2008) Treatment of refractory status epilepticus: literature review and a proposed protocol. Pediatric Neurology 38, 377–390.
Abend, N.S., Dlugos, D.J., Hahn, C.D., Hirsch, L.J. and Herman, S.T. (2010) Use of EEG monitoring and management of non-convulsive seizures in critically ill patients: a survey of neurologists. Neurocritical Care 12, 382–389.
Aiguabella, M., Falip, M., Villanueva, V., De La Pena, P., Molins, A., Garcia-Morales, I., Saiz, R.A., Pardo, J., Tortosa, D., Sansa, G. and Miro, J. (2011) Efficacy of intravenous levetiracetam as an add-on treatment in status epilepticus: a multicentric observational study. Seizure 20, 60–64.
Alldredge, B.K. and Lowenstein, D.H. (1999) Status epilepticus: new concepts. Current Opinion in Neurology 12, 183–190.
Arrol, L., Penderis, J., Garosi, L., Cripps, P., Gutierrez-Quintana, R. and Goncalves, R. (2012) Aetiology and long-term outcome of juvenile epilepsy in 136 dogs. Veterinary Record 170, 335.
Bateman, S.W. and Parent, J.M. (1999) Clinical findings, treatment, and outcome of dogs with status epilepticus or cluster seizures: 156 cases (1990–1995). Journal of the American Veterinary Medical Association 215, 1463–1468.
Beyreuther, B.K., Freitag, J., Heers, C., Krebsfanger, N., Scharfenecker, U. and Stohr, T. (2007) Lacosamide: a review of preclinical properties. CNS Drug Reviews 13, 21–42.
Bleck, T.P. (2005) Refractory status epilepticus. Current Opinion in Critical Care 11, 117–120.
Brandt, C., Heile, A., Potschka, H., Stoehr, T. and Loscher, W. (2006) Effects of the novel antiepileptic drug lacosamide on the development of amygdala kindling in rats. Epilepsia 47, 1803–1809.
Brauer, C., Jambroszyk, M. and Tipold, A. (2011) Metabolic and toxic causes of canine seizure disorders: A retrospective study of 96 cases. Veterinary Journal 187, 272–275.
Brophy, G.M., Bell, R., Claassen, J., Alldredge, B., Bleck, T.P., Glauser, T., Laroche, S.M., Riviello, J.J., Jr, Shutter, L., Sperling, M.R., Treiman, D.M., Vespa, P.M. and Neurocritical Care Society Status Epilepticus Guideline Writing (2012) Guidelines for the evaluation and management of status epilepticus. Neurocritical Care 17, 3–23.
Claassen, J., Hirsch, L.J., Emerson, R.G. and Mayer, S.A. (2002a) Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia 43, 146–153.
Claassen, J., Lokin, J.K., Fitzsimmons, B.F., Mendelsohn, F.A. and Mayer, S.A. (2002b) Predictors of functional disability and mortality after status epilepticus. Neurology 58, 139–142.
Claassen, J., Mayer, S.A., Kowalski, R.G., Emerson, R.G. and Hirsch, L.J. (2004) Detection of electrographic seizures with continuous EEG monitoring in critically ill patients. Neurology 62, 1743–1748.
Crandell, D.E. and Weinberg, G.L. (2009) Moxidectin toxicosis in a puppy successfully treated with intravenous lipids. Journal of Veterinary Emergency and Critical Care (San Antonio) 19, 181–186.
Eagleson, J.S., Platt, S.R., Strong, D.L., Kent, M., Freeman, A.C., Nghiem, P.P., Zheng, B. and White, C.A. (2012) Bioavailability of a novel midazolam gel after intranasal administration in dogs. American Journal of Veterinary Research 73, 539–545.
Frey, H.H. and Loscher, W. (1985) Pharmacokinetics of anti-epileptic drugs in the dog: a review. Journal of Veterinary Pharmacology and Therapeutics 8, 219–233.
Garzon, E., Fernandes, R.M. and Sakamoto, A.C. (2003) Analysis of clinical characteristics and risk factors for mortality in human status epilepticus. Seizure 12, 337–345.
Hardy, B.T., Patterson, E.E., Cloyd, J.M., Hardy, R.M. and Leppik, I.E. (2012) Double-masked, placebo-controlled study of intravenous levetiracetam for the treatment of status epilepticus and acute repetitive seizures in dogs. Journal of Veterinary Internal Medicine 26, 334–340.
Hasegawa, D., Orima, H., Fujita, M., Nakamura, S., Takahashi, K., Ohkubo, S., Igarashi, H. and Hashizume, K. (2003) Diffusion-weighted imaging in kainic acid-induced complex partial status epilepticus in dogs. Brain Research 983, 115–127.
Hasegawa, D., Matsuki, N., Fujita, M., Ono, K. and Orima, H. (2004) Kinetics of glutamate and gamma-aminobutyric acid in cerebrospinal fluid in a canine model of complex partial status epilepticus induced by kainic acid. Journal of Veterinary Medical Science 66, 1555–1559.
Hassell, T.M., Maguire, J.H., Cooper, C.G. and Johnson, P.T. (1984) Phenytoin metabolism in the cat after long-term oral administration. Epilepsia 25, 556–563.
Hirsch, L.J. (2004) Continuous EEG monitoring in the intensive care unit: an overview. Journal of Clinical Neurophysiology 21, 332–340.
Holopainen, I.E. (2008) Seizures in the developing brain: cellular and molecular mechanisms of neuronal damage, neurogenesis and cellular reorganization. Neurochemistry International 52, 935–947.
Huff, J.S. and Fountain, N.B. (2011) Pathophysiology and definitions of seizures and status epilepticus. Emerging Medicine Clinics of North America 29, 1–13.
Hulsmeyer, V., Zimmermann, R., Brauer, C., Sauter-Louis, C. and Fischer, A. (2010) Epilepsy in Border Collies: clinical manifestation, outcome, and mode of inheritance. Journal of Veterinary Internal Medicine 24, 171–178.
Jordan, K.G. (1995) Neurophysiologic monitoring in the neuroscience intensive care unit. Neurologic Clinics 13, 579–626.
Jull, P., Risio, L.D., Horton, C. and Volk, H.A. (2011) Effect of prolonged status epilepticus as a result of intoxication on epileptogenesis in a UK canine population. Veterinary Record 169, 361.
Knake, S., Hamer, H.M. and Rosenow, F. (2009) Status epilepticus: a critical review. Epilepsy and Behavior 15, 10–14.
Lado, F.A., Laureta, E.C. and Moshe, S.L. (2002) Seizure-induced hippocampal damage in the mature and immature brain. Epileptic Disorders 4, 83–97.
Lees, G., Stohr, T. and Errington, A.C. (2006) Stereoselective effects of the novel anticonvulsant lacosamide against 4-AP induced epileptiform activity in rat visual cortex in vitro. Neuropharmacology 50, 98–110.
Legriel, S., Azoulay, E., Resche-Rigon, M., Lemiale, V., Mourvillier, B., Kouatchet, A., Troche, G., Wolf, M., Galliot, R., Dessertaine, G., Combaux, D., Jacobs, F., Beuret, P., Megarbane, B., Carli, P., Lambert, Y.,
Pathophysiology and Management of Status Epilepticus
Bruneel, F. and Bedos, J.P. (2010) Functional outcome after convulsive status epilepticus. Critical Care Medicine 38, 2295–2303.
Leppik, I.E., Patterson, E.N., Coles, L.D., Craft, E.M. and Cloyd, J.C. (2011) Canine status epilepticus: a translational platform for human therapeutic trials. Epilepsia 52(Suppl. 8), 31–34.
Lowenstein, D.H. (1999) Status epilepticus: an overview of the clinical problem. Epilepsia 40(Suppl. 1), S3–8; discussion S21–22.
Lowenstein, D.H. (2005) Treatment options for status epilepticus. Current Opinion in Pharmacology 5, 334–339.
Lowenstein, D.H. (2006) The management of refractory status epilepticus: an update. Epilepsia 47(Suppl. 1), 35–40.
Lowenstein, D.H. and Alldredge, B.K. (1998) Status epilepticus. New England Journal of Medicine 338, 970–976.
Lowenstein, D.H. and Cloyd, J. (2007) Out-of-hospital treatment of status epilepticus and prolonged seizures. Epilepsia 48 (Suppl. 8), 96–98.
Mahmoudian, T. and Zadeh, M.M. (2004) Comparison of intranasal midazolam with intravenous diazepam for treating acute seizures in children. Epilepsy and Behavior 5, 253–255.
Marik, P.E. (2004) Propofol: therapeutic indications and side-effects. Current Pharmaceutical Design 10, 3639–3649.
Mazarati, A.M. and Wasterlain, C.G. (1999) N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neuroscience Letters 265, 187–190.
Mazarati, A.M., Baldwin, R.A., Sankar, R. and Wasterlain, C.G. (1998) Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Research 814, 179–185.
Mellema, L.M., Koblik, P.D., Kortz, G.D., Lecouteur, R.A., Chechowitz, M.A. and Dickinson, P.J. (1999) Reversible magnetic resonance imaging abnormalities in dogs following seizures. Veterinary Radiology and Ultrasound 40, 588–595.
Miro, J., Toledo, M., Santamarina, E., Ricciardi, A.C., Villanueva, V., Pato, A., Ruiz, J., Juvany, R. and Falip, M. (2012) Efficacy of intravenous lacosamide as an add-on treatment in refractory status epilepticus: A multicentric prospective study. Seizure 22(1), 72–79.
Mirsattari, S.M., Sharpe, M.D. and Young, G.B. (2004) Treatment of refractory status epilepticus with inhalational anesthetic agents isoflurane and desflurane. Archives of Neurology 61, 1254–1259.
Misra, U.K., Kalita, J. and Maurya, P.K. (2012) Levetiracetam versus lorazepam in status epilepticus: a randomized, open labeled pilot study. Journal of Neurology 259, 645–648.
Monteiro, R., Adams, V., Keys, D. and Platt, S.R. (2012) Canine idiopathic epilepsy: prevalence, risk factors and outcome associated with cluster seizures and status epilepticus. Journal of Small Animal Practice 53, 526–530.
Novy, J. and Rossetti, A.O. (2010) Oral pregabalin as an add-on treatment for status epilepticus. Epilepsia 51, 2207–2210.
Novy, J., Logroscino, G. and Rossetti, A.O. (2010) Refractory status epilepticus: a prospective observational study. Epilepsia 51, 251–256.
Overduin, L.M., Van Gogh, H., Mol, J.A. and Van Nes, J.J. (1989) Pharmacokinetics of three formulations of diphenylhydantoin in the dog. Research in Veterinary Science 46, 271–273.
Pakozdy, A., Leschnik, M., Tichy, A.G. and Thalhammer, J.G. (2008) Retrospective clinical comparison of idiopathic versus symptomatic epilepsy in 240 dogs with seizures. Acta Veterinaria Hungarica 56, 471–483.
Pakozdy, A., Leschnik, M., Sarchahi, A.A., Tichy, A.G. and Thalhammer, J.G. (2010) Clinical comparison of primary versus secondary epilepsy in 125 cats. Journal of Feline Medicine and Surgery 12, 910–916.
Pekcec, A., Unkruer, B., Stein, V., Bankstahl, J.P., Soerensen, J., Tipold, A., Baumgartner, W. and Potschka, H. (2009) Over-expression of P-glycoprotein in the canine brain following spontaneous status epilepticus. Epilepsy Research 83, 144–151.
Platt, S.R. and Haag, M. (2002) Canine status epilepticus: a retrospective study of 50 cases. Journal of Small Animal Practice 43, 151–153.
Raith, K., Steinberg, T. and Fischer, A. (2010) Continuous electroencephalographic monitoring of status epilepticus in dogs and cats: 10 patients (2004–2005). Journal of Veterinary Emergency and Critical Care (San Antonio) 20, 446–455.
Rajasekaran, K., Zanelli, S.A. and Goodkin, H.P. (2010) Lessons from the laboratory: the pathophysiology, and consequences of status epilepticus. Seminars in Pediatric Neurology 17, 136–143.
Rossetti, A.O. (2010) Treatment options in the management of status epilepticus. Current Treatment Options in Neurology 12, 100–112.
Rossetti, A.O. and Lowenstein, D.H. (2011) Management of refractory status epilepticus in adults: still more questions than answers. Lancet Neurology 10, 922–930.
Rossetti, A.O., Hurwitz, S., Logroscino, G. and Bromfield, E.B. (2006) Prognosis of status epilepticus: role of aetiology, age, and consciousness impairment at presentation. Journal of Neurology, Neurosurgery, and Psychiatry 77, 611–615.
Saito, M., Munana, K.R., Sharp, N.J. and Olby, N.J. (2001) Risk factors for development of status epilepticus in dogs with idiopathic epilepsy and effects of status epilepticus on outcome and survival time: 32 cases (1990–1996). Journal of the American Veterinary Medical Association 219, 618–623.
Schriefl, S., Steinberg, T.A., Matiasek, K., Ossig, A., Fenske, N. and Fischer, A. (2008) Etiologic classification of seizures, signalment, clinical signs, and outcome in cats with seizure disorders: 91 cases (2000–2004). Journal of the American Veterinary Medical Association 233, 1591–1597.
Serrano, S., Hughes, D. and Chandler, K. (2006) Use of ketamine for the management of refractory status epilepticus in a dog. Journal of Veterinary Internal Medicine 20, 194–197.
Shorvon, S. and Ferlisi, M. (2012) The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain 135, 2314–2328.
Silbergleit, R., Durkalski, V., Lowenstein, D., Conwit, R., Pancioli, A., Palesch, Y., Barsan, W. and Investigators, N. (2012) Intramuscular versus intravenous therapy for prehospital status epilepticus. New England Journal of Medicine 366, 591–600.
Smith, R.D. and Lomas, T.E. (1978) Modification of cardiovascular responses to intravenous phenytoin by dopa-mine in dogs: evidence against an adverse interaction. Toxicology and Applied Pharmacology 45, 665–673.
Stohr, T., Kupferberg, H.J., Stables, J.P., Choi, D., Harris, R.H., Kohn, H., Walton, N. and White, H.S. (2007) Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Research 74, 147–154.
Swisher, C.B., Doreswamy, M., Gingrich, K.J., Vredenburgh, J.J. and Kolls, B.J. (2012) Phenytoin, levetiracetam, and pregabalin in the acute management of refractory status epilepticus in patients with brain tumors. Neurocritical Care 16, 109–113.
Swisher, C.B., Doreswamy, M. and Husain, A.M. (2013) Use of pregabalin for nonconvulsive seizures and nonconvulsive status epilepticus. Seizure 22, 116–118.
Thomas, W.B. (2010) Idiopathic epilepsy in dogs and cats. Veterinary Clinics of North America - Small Animal Practice 40, 161–179.
Trinka, E., Hofler, J. and Zerbs, A. (2012) Causes of status epilepticus. Epilepsia 53(Suppl. 4), 127–138.
Vasile, B., Rasulo, F., Candiani, A. and Latronico, N. (2003) The pathophysiology of propofol infusion syndrome: a simple name for a complex syndrome. Intensive Care Medicine 29, 1417–1425.
Vespa, P.M., Nuwer, M.R., Nenov, V., Ronne-Engstrom, E., Hovda, D.A., Bergsneider, M., Kelly, D.F., Martin, N.A. and Becker, D.P. (1999) Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. Journal of Neurosurgery 91, 750–760.
Vespa, P.M., Miller, C., Mcarthur, D., Eliseo, M., Etchepare, M., Hirt, D., Glenn, T.C., Martin, N. and Hovda, D. (2007) Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Critical Care Medicine 35, 2830–2836.
Vulliemoz, S., Perrig, S., Pellise, D., Vargas, M.I., Gasche, Y., Ives, J.R. and Seeck, M. (2009) Imaging compatible electrodes for continuous electroencephalogram monitoring in the intensive care unit. Journal of Clinical Neurophysiology 26, 236–243.
Wallace, S.J. (1997) Nasal benzodiazepines for management of acute childhood seizures? Lancet 349, 222.
Wasterlain, C.G., Stohr, T. and Matagne, A. (2011) The acute and chronic effects of the novel anticonvulsant lacosamide in an experimental model of status epilepticus. Epilepsy Research 94, 10–17.
Weissl, J., Hulsmeyer, V., Brauer, C., Tipold, A., Koskinen, L.L., Kyostila, K., Lohi, H., Sauter-Louis, C., Wolf, M. and Fischer, A. (2012) Disease progression and treatment response of idiopathic epilepsy in Australian Shepherd dogs. Journal of Veterinary Internal Medicine 26, 116–125.
Wu, Y.W., Shek, D.W., Garcia, P.A., Zhao, S. and Johnston, S.C. (2002) Incidence and mortality of generalized convulsive status epilepticus in California. Neurology 58, 1070–1076.
Yas-Natan, E., Segev, G. and Aroch, I. (2007) Clinical, neurological and clinicopathological signs, treatment and outcome of metaldehyde intoxication in 18 dogs. Journal of Small Animal Practice 48, 438–443.
Zimmermann, R., Hulsmeyer, V., Sauter-Louis, C. and Fischer, A. (2009a) Status epilepticus and epileptic seizures in dogs. Journal of Veterinary Internal Medicine 23, 970–976.
Zimmermann, R., Hulsmeyer, V.I., Sauter-Louis, C. and Fischer, A. (2009b) Status epilepticus and epileptic seizures in dogs. Journal of Veterinary Internal Medicine 23, 970–976.
Zimmermann, R., Steinberg, T.A., Raith, K., Hulsmeyer, V. and Fischer, A. (2010) Canine status epilepticus due to acute intoxication. Tierarztl Prax Ausg K Kleintiere Heimtiere 38, 285–294.
25 Novel and Adjunctive Treatments
Simon Platt BVM&S MRCVS Dipl. ACVIM (Neurology) Dipl. ECVN
Professor Neurology and Neurosurgery Service, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, Georgia, USA
Newly available anti-epileptic medications hold promise for people and veterinary patients with intractable epilepsy (Martlé et al., 2013). However, at least 25–50% of human patients with epilepsy continue to experience seizures or debilitating side-effects from medication (Schachter and Saper, 1998). Around 75–85% of dogs with idiopathic epilepsy will continue to have seizures (Heynold et al., 1997; Berendt et al., 2002, 2007; Arrol et al., 2012) and around 20–30% will remain poorly controlled (<50% reduction of seizure frequency) despite adequate treatment with phenobarbital (PB) and/or potassium bromide (KBr) (Schwartz-Porsche et al., 1985; Podell and Fenner, 1993; Trepanier et al., 1998). Therefore there is a need for additional therapies, which can be used as an adjunct for those patients with an unsatisfactory response to the standard medicinal approach.
This chapter reviews some of the adjunct, sometimes termed alternative, treatments which are being attempted in both humans and dogs and cats. These include some treatments that have undergone testing in controlled trials (e.g. vagus nerve stimulation), some treatments that we are just starting to understand although they have been around for almost a century (e.g. dietary therapy) and others that have not received much attention. In some patients, which remain uncontrolled by anti-epileptic medication, the use of these less conventional approaches may offer more than just hope. The theoretical benefits of these approaches, which include vagus nerve stimulation, dietary therapy, acupuncture as well as homeopathy, and the clinical information on their use where available is presented. However, there is very limited information available on the use of any of these therapies in veterinary medicine and so caution needs to be taken in interpretation of the human literature or translation of the potential benefit seen in experimental animal models.
Neurostimulation
Vagus nerve stimulation (VNS) is currently the only approved non-pharmacological treatment in the USA for seizure therapy in humans. However, multiple forms of stimulation have been proposed at sites as varied as cerebellum, anterior thalamus, centromedian thalamus, subthalamic nucleus, hippocampus, caudate, locus coeruleus, corpus callosum, mammillothalamic tract and the cortical seizure focus (Lockman and Fisher, 2009).
Vagus nerve stimulation
Up to 90% of the vagus nerve consists of afferent fibres originating in the viscera and
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
predominantly terminating in the brainstem (nucleus of the solitary tract) (Fornai et al., 2011). From here, signals are projected to multiple cortical and subcortical regions.
In the early part of last century, it was found that stimulation of the cervical portion of the vagus nerve in cats altered cortical electrical activity; furthermore, electrical stimulation of the vagus nerve at high frequencies and intensities caused ‘desynchronization’ of cortical neuronal activity (Zanchetti et al., 1952; Chase et al., 1967). Studies in dogs, rats and monkeys subsequently revealed that intermittent stimulation of the left cervical vagus trunk could effectively prevent experimentally induced seizures (Lockard et al., 1990; Woodbury and Woodbury, 1990; Zabara, 1992).
VNS was approved in Europe in 1994 and in the USA and Canada in 1997 for therapy of epilepsy in humans, based upon pivotal trials in patients with focal and secondarily generalized seizures in patients over 12 years of age (Ben-Menachem et al., 1994; Handforth et al., 1998; Fisher and Handforth, 1999). VNS has been shown to be effective for focal and secondarily generalized seizures in paediatric populations and some trials have suggested efficacy for some generalized seizures (Labar et al., 1999; Elliott et al., 2011).
In clinical human use, VNS requires a team of health-care providers to implant the device, teach the patient and his or her family about its use, titrate the stimulation parameters to optimum clinical response, and monitor the patient’s side-effects and the device’s battery life. The device or implant is a flat, round piece of metal that measures about an inch and a half (4 cm) across and 10–13 mm thick, depending on the model used. Newer models may be somewhat smaller. The stimulator contains a battery, which can last from 1 to 15 years. When the battery is low the stimulator needs to be replaced via a surgical procedure that is similar to but less involved than the original implantation.
The surgical implantation procedure usually takes about 50 to 90 min with the patient under general anaesthesia, and in such cases a hospital stay of one night may be required. However, some physicians perform the procedure using local anaesthesia on an outpatient basis. As with all surgeries, there is a small risk of infection. Other surgical risks of VNS include inflammation or pain at the incision site, damage to nearby nerves and nerve constriction. The procedure requires two small incisions. The first is made on the upper left side of the chest where the pulse generator is implanted. A second incision is made horizontally on the left side of the lower neck, along a crease of skin. This is where the thin, flexible wires that connect the pulse generator to the vagus nerve are inserted. Because the right vagus innervates the cardiac atria more than the left vagus nerve and the left vagus nerve provides the predominant innervation of the ventricles, electrical stimulation of the left vagus nerve has generally been used in clinical practice, though right-sided VNS has been reported safe in one case series and is equally effective against seizures as left-sided stimulation in rat models of epilepsy (Krahl et al., 2003; McGregor et al., 2005).
The stimulator is generally activated 2 to 4 weeks after implantation. The doctor programs the stimulator with a small hand-held computer, programming software and a programming wand. The strength and duration of the electrical impulses are programmed. The doctor can program the output current (typically 1mA), signal frequency (typically 30 Hz), signal pulse width (typically 500 ms), signal on-time (typically 30 s) and signal off-time (typically 5 min). In addition, magnet-activated stimulus parameters – pulse width, output current and on-time – are also programmable. Patients are provided with a handheld magnet to control the stimulator at home (which must be activated by the doctor to magnet mode). When the magnet is placed over the pulse generator site and then moved away, extra stimulation is delivered, regardless of the treatment schedule. Holding the magnet over the pulse generator will turn the stimulation off. Removing it will resume the stimulation cycle. This can be done by the patient, family members, friends or caregivers.
The stimulator settings employed for on-demand stimulation usually utilize a higher current and pulse width than those used for intermittent stimulation. Some patients have reported that on-demand stimulation interrupts a seizure or reduces its severity if administered at the onset of the seizure, and can be used by patients to attempt to abort an ongoing seizure (Boon et al., 2001).
Mechanism of Action
The way VNS exerts its antiseizure effect remains uncertain. Early work showed that repetitive VNS either synchronizes or desynchronizes cortical activity in anesthetized animals, depending on stimulus frequency and current strength, which determines whether myelinated fibres are activated (Chase et al., 1967; Chase and Nakamura, 1968). Desychronization of cortical rhythms implied a possible anticonvulsant effect of VNS during this early work, however, more recently, similar studies have been unable to find a relationship between clinical response to VNS and a reduction of electroencephalographic (EEG) discharges after implantation compared to pre-implantation of the device (Ebus et al., 2004). Additionally, human studies have revealed that VNS induces little if any effect on EEG background rhythms (Hammond et al., 1992; Salinsky and Burchiel, 1993). That the sole anti-epileptic mechanism of VNS is to desynchronize cortical activity should be considered unlikely, as this effect would only last during the stimulation time of the vagus nerve. However, VNS is undertaken on an intermittent basis and so there must be a more continuous or longer lasting effect taking place.
Once the vagus nerve is peripherally stimulated, the major sensory nucleus that receives vagal afferents is the nucleus of the solitary tract (NST); it is likely that NST may be key in bridging VNS to central anti-epileptic circuitries (Fornai et al., 2011). The NST projects to the amygdala, cerebellum, hypothalamus, thalamus, parabrachial nucleus, raphe nuclei and the nucleus of the locus coeruleus (LC). The LC has been a major focus of work investigating the anti-epileptic effect of VNS. Permanent loss or functional inactivation of LC neurons makes the epileptic activity refractory to the effects of VNS (Krahl et al., 1998). Additionally, it has been shown that the LC is important in both limiting the spread and duration of a seizure as damage to LC converts sporadic seizures into long-lasting, self-sustaining status epilepticus (Giorgi et al., 2006). However, other brainstem structures, such as the dorsal raphe nucleus, are involved, as the anticonvulsant effects of VNS are not completely lost by inactivation of LC neurons (Fornai et al., 2011). In keeping with the influence of the NST on LC neurons, there is a significant increase in norepinephrine (NE) release during VNS (Roosevelt et al., 2006). The LC represents the largest group of NE neurons in the human brain (Ruffoli et al., 2011). The impact of the LC and this NE release in reducing epileptic activity has been shown in a variety of studies (Szot et al., 1999; Giorgi et al., 2006; Chachua et al., 2010). In order to produce a modulation of epilepsy, which is an exclusively cortical phenomenon, LC is expected to produce final effects on the cerebral cortex and it does so by giving rise to axons, which directly enter the cortex and possess rich collateral branching, enabling a variety of cerebral lobes and gyri to be targeted (Ruffoli et al., 2011). The projections rising from the LC do not need to relay within the thalamic nuclei and produce a faster effect on EEG activity. Interestingly, these projections possess different varicosities (Giorgi et al., 2004), which determine a marked paracrine influence on innervated structures.
Electrical stimulation of the peripheral vagus nerve probably requires synaptic transmission to mediate antiseizure activity. Regional alterations in synaptic activity cause rapid changes in regional cerebral blood flow (rCBF). It has been found that VNS acutely increases rCBF at sites that receive vagal afferents and higher-order projections, including dorsal medulla, somatosensory cortex (contralateral to stimulation), thalamus and cerebellum bilaterally, and several limbic structures (including the hippocampus and amygdala bilaterally) (Henry et al., 1999). The extent of bilateral thalamic changes in blood flow has been correlated with reductions in seizure frequency based on positron emission tomography (PET) studies (Henry et al., 1999).
Efficacy
The first human patient with epilepsy was treated with VNS in 1988 (Penry and Dean, 1990), followed by two pivotal trials of human patients with focal epilepsy, which were blinded and randomized comparing two different VNS stimulation protocols: high stimulation (30 Hz, 30 s on, 5 min off, 500 µs pulse width) and low stimulation (1 Hz, 30 s on, 90 to 180 min off, 130 µs pulse width). Enrolled patients were at least 12 years of age, with at least six seizures per month treated with a mean of 2.1 anti-epileptic medications (AEMs) at the start of the study (Ben-Menachem et al., 1994; George et al., 1994; Vagus Nerve Stimulation Study Group, 1995; Handforth et al., 1998).
In both studies, the primary measure of efficacy was the percentage change in seizure frequency during VNS treatment compared to the pre-implantation baseline. Changes in seizure frequencies in the high- and low-stimulation groups were then compared in each study. The hypothesis was that the low-stimulation treatment was less effective than the high-stimulation treatment. The high-stimulation group had a mean reduction in seizure frequency of 24.5–28% versus 6.1–15% for the low-stimulation group (p = 0.01). Furthermore, 31% of patients receiving high stimulation had at least 50% reduction in seizures compared to 13% of patients in the low-stimulation group (p = 0.02). Thus, high stimulation was more effective than low.
Seizure control is maintained in long- term studies of VNS, including childhood epilepsy, but these results should be interpreted cautiously since long-term studies are not blinded, and stimulation parameters and AEM dosages could be adjusted as clinically indicated (Michael et al., 1993; George et al., 1994; Salinsky et al., 1996; DeGiorgio et al., 2000; Uthman et al., 2004; Vonck et al., 2004; Huf et al., 2005; Janszky et al., 2005). One retrospective study of 269 human patients whose AEMs were kept constant for 1 year after VNS implantation found a 45% seizure reduction at 3 months post-implantation compared to pre-implantation baseline, and 58% reduction at 12 months post-implantation (Labar et al., 1999).
The use of VNS as a means of seizure control has been evaluated in dogs. Preclinical studies were performed in laboratory dogs to obtain preliminary data on the effectiveness of VNS in controlling seizures and establish optimal stimulation parameters for its use (Zabara, 1992). In these studies, seizures were induced in anaesthetized dogs by IV administration of either strychnine or pentylenetetraxol, and repetitive stimulation of the cervical portion of the vagus nerve caused termination of seizure activity
A clinical canine study was performed in which the purpose was to evaluate the efficacy of VNS in controlling medically refractory seizures and determine the potential safety and tolerability of the surgically implanted VNS device in epileptic dogs (Muñana et al., 2002). Dogs included in the study had to have a 1 year documented history of seizures; a historical seizure frequency of at least 5 seizures/month and a seizure-free period of no longer than 2 weeks, or cluster seizures occurring at least once per month despite treatment with therapeutic serum concentrations of orally administered AEMs. A programmable vagal nerve stimulator delivering intermittent stimulation to the left cervical trunk of the vagus was surgically implanted in each of ten dogs. The clinical trial was designed as a double-masked crossover study with each dog undergoing a treatment period during which the device was active and a control period in which the device was inactive. Dogs were assigned randomly to two 13-week test periods, one with nerve stimulation and one without nerve stimulation. Owners recorded data on seizure frequency, duration and intensity, as well as adverse effects. No significant difference in seizure frequency, duration or severity was detected between overall 13-week treatment and control periods. However, during the final 4 weeks of the treatment period, a significant decrease in mean seizure frequency (34.4%) was detected, compared with the control period. This difference may reflect an increase in the efficacy of VNS over time. The effectiveness of VNS in controlling seizures improves for up to 24 months after initiation of treatment in human epileptics, and it is possible that dogs respond similarly (Muñana et al., 2002).
Side effects and safety
The side effects of VNS for epilepsy in humans include hoarseness (37%), throat pain (11%), coughing (7%), shortness of breath (6%), tingling (6%) and muscle pain (6%) (Ramsay et al., 1994). These effects are common but not dangerous. VNS has no known adverse cardiac complications in patients with epilepsy apart from the rare (0.1% of cases) documentation of ventricular asystole intraoperatively during initial testing for implantation of the vagus nerve stimulator (Ramsay et al., 1994). Continuous ECG monitoring intraoperatively before generator implantation is now recommended. In large controlled studies, no autonomic dysfunction, including significant changes in mean heart rate, heart rate variability or bradycardia, was noted despite Holter monitoring and serial general physical examinations (Handforth et al., 1998). Left-sided rather than right VNS has been suggested to minimize the arrhythmogenic effects (Schachter and Saper, 1998; Tatum et al., 1999). Among 444 human patients who continued VNS after participating in a clinical study, the most commonly reported side-effects at the end of the first year post-implantation were voice alteration (29%) and tingling (12%), at the end of 2 years, voice alteration (19%) and cough (6%) and at 3 years, shortness of breath (3%) (Morris and Mueller, 1999). No adverse effects were seen in the short-term canine study but complications included transient bradycardia, asystole and apnea during intraoperative device testing, and seroma formation, subcutaneous migration of the generator and transient Horner’s syndrome during the 14-day period between surgery and suture removal (Muñana et al., 2002). The higher incidence of bradycardia seen in the dogs was expected because of anatomic differences between humans and dogs (Muñana et al., 2002). In humans, care is taken to wrap the electrodes around the left vagus nerve at a point distal to where most of the cardiac branches leave the nerve. In dogs, the cardiac branches leave the nerve at a more distal point within the thoracic cavity. Consequently, it is not possible to spare the cardiac branches from the effects of direct stimulation. Previous studies on the cardiac effects of VNS revealed that stimulation of the cervical portion of the vagus nerve in anaesthetized dogs consistently induced bradycardia or asystole (Asconape et al., 1999). However, there were no changes in cardiac rate or rhythm in dogs when they were evaluated in a non-anaesthetized state (Munana et al., 2002).
Non-invasive vagus nerve stimulation
Although implanted VNS has shown promise in the treatment of epilepsy, the morbidity associated with the invasive implantation procedure along with the cost of these devices, limit the potential for widespread and frequent use. To address this situation, ElectroCore® developed a proprietary electrode configuration and signal that allowed for biologically active electrical fields to be delivered to the vagus nerve without the nociceptive skin pain associated with standard TENS devices. The device in use (GammaCore®) is not implantable, is not in direct physical contact with the vagus nerve and only provides stimulation for a maximum of 120 s per use. Additionally, the stimulation intensity can be controlled by the patient or, in veterinary medicine, the owner.
The signal characteristics and the electrical field amplitude at the location of the vagus nerve are similar between the GammaCore device and implanted VNS devices (data on file at ElectroCore®). Non-invasive VNS has been successfully investigated for its clinical effect in migraines using an accepted animal (rat) model of recurrent headaches (Oshinsky and Gomonchareonsiri, 2007). It has been shown to reduce extracellular trigeminal nucleus caudalis glutamate concentrations in comparison to untreated rats. Such a positive biochemical outcome has given rise to the possibility that non-invasive VNS may be equally effective for seizure activity although the clinical trials are currently underway.
There is other recent evidence in the literature of transcutaneous VNS for seizure treatment which points toward a more acceptable and affordable manner in which treatment can be undertaken (Dietrich et al., 2008; Yang et al., 2011).
Transcranial magnetic brain stimulation
Electrical shocks to the scalp can activate cortical neurons, but the stimulation tends to hurt (Merton and Morton, 1980); transcutaneous magnetic stimulation (TMS) is less painful. Magnetic field-induced brain currents fall off rapidly with distance from the magnetic stimulator coil, so great efforts have been made to produce coils that can stimulate focally and relatively deeply into brain tissue (Theodore and Fisher, 2004; Deng et al., 2008). Figure-of-eight coils are widely in use by virtue of these characteristics.
Early case series of TMS for epilepsy generally were favourable (Theodore and Fisher, 2004). Nine human patients with focal or secondarily generalized seizures, two from temporal and seven from extra-temporal regions were given TMS (Tergau et al., 1999). A round magnetic coil stimulated the vertex head region at one pulse every 3 s for two trains of 500 pulses per day. Weekly seizure frequency declined from 10.3 ± 6.6 before stimulation to 5.8 ± 6.4, significant at p = 0.048. Subsequent case series of TMS showed benefit for seizures in some and little or no benefit in others (Wassermann and Lisanby, 2001; Morales et al., 2005; Rotenberg et al., 2008; Rossi et al., 2009). Positioning of the stimulating coil over the seizure focus might be important in determination of success, according to one study that compared vertex stimulation to targeted TMS (Daniele et al., 2003).
Three controlled trials of TMS for epilepsy have been accomplished. In a positive trial, Fregni et al. (2006) targeted TMS to sites of cortical dysplasia in 21 patients with medication-resistant epilepsy. Patients were subjected to five consecutive daily 20 min sessions of stimulation at one per second using either a figure-of-eight real stimulation coil or a fabricated coil looking and sounding similar to a real coil but delivering no stimulation. The epileptogenic focus was targeted as the site of stimulation, except in four patients with diffuse abnormalities, in whom stimulation was delivered to the vertex. By 2, 4 and 8 weeks after stimulation, seizure frequency was reduced respectively to 72%, 53% and 58% of baseline, each of which was statistically significant. EEG epileptiform discharges also were reduced. Two other controlled studies were negative. Cantello et al. (2007) stimulated 43 patients with medication-resistant predominantly focal cortical epilepsies. After a 12-week baseline TMS was initiated via two stacked stimulating coils over the vertex. Active treatment was stimulation with the one near the scalp, and sham with stimulation by the upper coil distant from the scalp. Stimulation was set at two daily series of 500 stimuli at
0.3 Hz, separated by a 30 s interval. The stimulus intensity was 100% of the motor-evoked threshold. Although the study showed trends in favour of stimulation, neither seizure frequency nor EEG epileptiform activity changed significantly with active versus sham stimulation. Theodore et al. (2002) evaluated TMS in 21 patients with localization-related epilepsy. TMS was given at 120% of the motor-evoked threshold at 1 pulse per second for 15 min twice daily for 1 week at 120%. The coil was positioned over the best estimate of the region of the seizure focus. Sham stimulation was given with the coil angled away from the head. The patients were then observed on a stable drug regimen for 2 months. Neither focal nor generalized seizures improved significantly with TMS active stimulation in comparison to sham stimulation. A trend toward short-term benefit was noted in patients with lateral temporal seizure foci, where magnetic fields would best penetrate the focus.
Experience collectively leaves open the question of effectiveness of TMS for epilepsy. One of three controlled studies showed efficacy. That study targeted stimulation to superficial regions of cortical dysplasia, which may have been a factor in efficacy. Other differences in stimulation parameters, such as frequency, intensity, duration of the train and other factors could have contributed to different study outcomes. In addition, compared to other trials of neurostimulation, TMS trials have stimulated only during a small fraction of each trial day.
Magnetic stimulation is not entirely benign, in that it inadvertently can instigate seizures, even with single pulses (Kratz et al., 2011). A systematic literature review found 16 cases of seizures with TMS (Rossi et al., 2009). A consensus conference on safety of TMS concluded that TMS was contraindicated when metallic hardware, such as a cochlear implant or medication pump, was in close proximity to the stimulation site (Rossi et al., 2009). Special care is required with untested stimulation parameters, patients with a seizure history or brain lesions or medications that lower seizure thresholds, or pregnancy or heart disease.
The author could find no evidence of TMS being performed in dogs for the treatment of epilepsy.
Thalamic stimulation
The first devices used to treat epilepsy were forms of electrical stimulation. Electrical stimulation to map human brain function may have started in 1884, when the Cincinnati surgeon Robert Bartholow observed contra-lateral movements with electrical stimulation of cortex during repair of cranial osteomyelitis (Gildenberg, 2005).
Deep brain electrical stimulation (DBS) to reduce seizures is credited to the New York neurosurgeon Irving Cooper, who reported improvement in seizure frequency with stimulation either of cerebellum or the anterior thalamus (Cooper et al., 1973, 1980). Cooper’s positive results were qualitative and uncontrolled with little detail on individual degrees of improvement and co-morbid conditions. In subsequent years, about a dozen uncontrolled studies showed benefit of cerebellar stimulation to treat epilepsy, but two small, blinded studies were negative (Krauss and Fisher, 1993). DBS for epilepsy fell out of favour for many years and came back to interest with the success of VNS for epilepsy and DBS for movement disorders. After cerebellum, centromedian thalamus was the primary target of stimulation, pioneered by the Velascos in Mexico City, but a small cross-over trial was negative (Velasco et al., 1987, 2006; Fisher et al., 1992).
A series of studies showed benefit of DBS of anterior thalamus in experimental models of epilepsy (Fisher, 2012; Graber and Fisher, 2012). Based upon promising animal experimentation and the early work of Cooper, six small unblinded trials of anterior nucleus stimulation for medication-resistant epilepsy were published, showing a conglomerate mean 47% reduction in seizures compared to baseline (Graves and Fisher, 2005). Uncontrolled stimulation studies are subject to several types of potential bias, including placebo effect, regression to the mean, micro-lesion effects from electrode placement and other unknown confounding factors. Therefore, Fisher et al. (2010) performed a randomized, placebo-controlled, multi-centre trial of anterior nucleus stimulation in patients with medication-resistant focal and secondarily generalized seizures, called SANTE, for stimulation of the anterior nucleus of thalamus for epilepsy. Randomization was performed on 110 patients either to 5 V or 0 V (placebo) stimulation of bilateral anterior nuclei of thalamus, at 145 pulses per second, 0.9 ms pulses referential to the stimulation case, with stimulation on for 1 min and off for 5 min. The group had a median of about 20 seizures per month and a mean of 57 seizures per month at baseline. Stimulation was begun 1 month after implantation of the deep-brain leads and continued for a 3-month blinded phase. Seizure frequency declined 20% in the month after implantation prior to initiation of electrical stimulation, either to nonspecific or micro-lesion effects. By the end of the blinded phase, the treated group continued to improve, to a median level 40.5% less than baseline, compared to only 14.5% in the 0 V group (p = 0.038). The control group received 5 V stimulation at the end of the blinded phase. Seizures declined over the next 2 months to levels encountered in the initially stimulated group. Improvement was sustained, with seizures in the terminal 3 months of stimulation at 3 years measuring a median 58% reduction compared to baseline. In the blinded phase, stimulation produced significant reduction in injuries due to seizures, frequency of complex focal (partial) seizures, seizures originating from the temporal lobes and seizures predesignated as ‘most severe’ by the patient. Responder rates for 50% improvement and quality of life did not significantly improve during the 3-month blinded phase, but did in the open-label and long-term follow-up stages from 1 to 3 years after implantation. In the long-term phase, 14% of patients became seizure-free for at least 6 months. Patients who previously had not benefited from VNS or epilepsy surgery had the same favourable response to DBS as did the overall group.
Complications of stimulation consisted of occasional chest pain or other paresthesias, need for repositioning leads and superficial infections. No symptomatic brain haemorrhages were seen, though neuroimaging showed asymptomatic blood in five patients. Neuropsychological tests showed no difference in cognitive or profiles of mood scores, but more stimulated patients reported symptoms of depression and memory impairment. Five patients had status epilepticus, two related to initiation of stimulation, and resolving with reduction of voltage. Rates of depression, status epilepticus, depression, suicide and sudden unexpected death in epilepsy (SUDEP) all were within the expected ranges for a population of people with refractory epilepsy.
The conclusion of the SANTE study was that stimulation of the anterior nuclei of thalamus reduced the number of seizures in patients with medication-resistant epilepsy. Complications were similar to those encountered with DBS for movement disorders, with additional concerns raised about possible subjective symptoms of depression and memory impairment.
Despite the positive effect of DBS, the author could find no evidence of this procedure being performed in dogs or cats with seizure activity.
Dietary Therapy
Essential fatty acid supplementation
Dietary intake of fatty acids (FA) affects the fatty acid composition of neuronal cell membrane phospholipids. FA can modulate electrical signal transduction mechanisms, by affecting ion channel functions and receptor systems (Haag, 2003). The omega-3 FAs, eicosapentaenoic acid (EPA, 20:5n-3) and docosahexaenoic acid (DHA, 22:6n-3), derived mainly from fish oil, are the ones incorporated into neuronal phospholipids. Pharmacology studies show that EPA and DHA applied extracellularly raise the stimulatory thresholds of CA1 neurons in hippocampal slices, and infusions of EPA and DHA are equipotent in raising seizure thresholds in the male Wistar rat (Voskuyl et al., 1998; Xiao and Li, 1999). Furthermore, prolonged oral administration of a-linolenic acid and linoleic acid in a 1:4 mixture protects rats from having seizures in four different epilepsy models (Yehuda et al., 1994).
Pharmacology studies also show that the omega-6 FA arachidonic acid (AA) inhibits sodium currents and synaptic transmission (Fraser et al., 1993). Hence AA may also have an impact on seizures.
Other studies demonstrate the role of inflammatory mediators in epilepsy: pro-inflammatory mediators are elevated in rodent models of epilepsy and in humans with epilepsy, whereas increasing anti-inflammatory mediators appear to have anticonvulsant effects (Naffah-Mazzacoratti et al., 1995; Vezzani et al., 2002; Hulkkonen et al., 2004). EPA reduces pro-inflammatory mediators by inhibiting phospholipase A2 and cyclooxygenase-2 enzymes and is itself metabolized to form anti-inflammatory prostaglandins (Finnen and Lovell, 1991; Obata et al., 1999). Increasing omega-3 FA intake has been shown to lower plasma inflammatory markers (Pischon et al., 2003). A minimum of 3 months of dietary fish oil supplementation was necessary to raise seizure thresholds in a recent study on rats, which also demonstrated that this effect was more likely mediated by DHA than EPA (Taha et al., 2013). Therefore there is some evidence from rodent studies that omega-3 FA may increase seizure thresholds. The studies on inflammatory markers also suggest that EPA can reduce these markers, which may lead to a reduced seizure frequency.
Clinically, Schlanger et al. (2002) reported on an open clinical trial in which five human patients took omega-3 FA supplements. Though the results were encouraging, it was not possible to draw any definitive conclusions. A double-blind placebo-controlled study in humans demonstrated a reduction in seizures within the first 6 weeks of starting supplementation with omega-3 FAs, both for the supplement group during the treatment phase (weeks 1–6) and for the placebo group during the maintenance phase (weeks 13–18). However, this reduction was not maintained in the subsequent 6-week periods (Yuen et al., 2005).
In dogs, a recent blinded, cross-over, placebo-controlled trial evaluating EFA was performed with two phases of 12 weeks (Matthews et al., 2012). During the first 12 weeks dogs were supplemented with an EFA product, followed by a 12-week phase of supplementation with a placebo. Fifteen dogs with idiopathic epilepsy were included with a seizure frequency between more than one per month and more than one per year, no magnetic resonance imaging (MRI) abnormalities, normal cerebrospinal fluid analysis and no clinical pathology abnormalities (including haematology, biochemistry and dynamic bile acid evaluation). Fourteen out of 15 dogs were treated with AEMs, which included phenobarbital and/ or potassium bromide at established therapeutic serum levels and the medication dosage was not changed during the trial. Analysis of seizure frequency showed that the number of seizures during the active supplementation and placebo phases was similar between the two periods; 9/13 dogs with full follow-up had the same seizure frequency (P = 0.1). Seizure severity was also similar between the two trial periods (P = 0.93). Of the dogs in which seizures were observed, the median change in severity was zero. Median difference in individual dogs in both frequency and severity between the two treatments was identical. Three dogs experienced adverse events potentially related to the active supplement (diarrhoea, skin irritation and halitosis). One dog experienced severe diarrhoea and therefore 2 weeks into the trial period the dose was reduced to one-third of the original dose. A subsequent increase in the dose led to recurrence of the diarrhoea, so the dog completed the 12-week trial period at the reduced dose. Although this was a well-constructed study, the duration of treatment may just have been too short to evaluate the efficacy of this supplementation and several of the dogs may not have had a high enough entrance seizure frequency for an effect to be seen.
Hypoallergenic diet
The possibility that certain foods or allergens may induce convulsions has been reported in the literature over the last century (Frediani et al., 2004). Multiple anecdotal websites exist documenting the potential role of dietary sensitivity in canine epilepsy, although there are no published, peer-reviewed reports. Most of the human studies have failed to show a close correlation between food allergy and epilepsy, being anecdotal and open to various aetiological hypotheses. One interesting publication describes three children with cryptogenic focal epilepsy, diagnosed by means of electroencephalography, with behavioural disorders (hyperactivity, sleep disorders and writing difficulties) (Pelliccia et al., 1999). In these patients, instead of using antiepileptic medications (AEMs), treatment was based upon a cow’s milk-free diet, working on the hypothesis that there could be a causal relationship between intolerance to this food item and the epileptic symptoms. An improvement was observed in the children’s behaviour and, moreover, the electroencephalographic anomalies disappeared. Upon double-blind oral provocation tests, these patients did not have an immediate reaction, taking a few days to show signs. Starting the controlled diet again led, in all cases, to disappearance of the electroencephalographic abnormalities.
Another report describes the consistent disappearance of focal idiopathic epilepsy symptoms in a 9-year-old child as a result of diet free of cow’s milk protein (Frediani et al., 2004). The case also appears to confirm the possible role of food allergy in certain types of epilepsy in human patients of paediatric age. Indeed, when compared to children without epilepsy, a significantly higher incidence of allergy to cow’s milk and asthma was documented in epileptic children (Frediani et al., 2001). Quite what this means for dogs which are likely to have minimal exposure to milk proteins is unknown. However, children with cow’s milk allergies can also be allergic to beef protein and cereals, giving rise to the thought that our veterinary patients may also have the potential to express sensitivity to foods as seizure activity (Martelli et al., 2002; Jarvinen et al., 2003). If this is the case, commercially available hypoallergenic diets used for the control of skin diseases in dogs and cats may have an adjunctive role in the control of seizure activity in some patients. However, controlled prospective studies are necessary before accurate dietary advice can be given.
Ketogenic diet
The ketogenic diet (KD) is a high-fat, low-carbohydrate, adequate protein diet that
has been used as an alternative therapy for intractable seizures since the early 1920s in humans (Kessler et al., 2011; Rho and Stafstrom, 2012; Martlé et al., 2013). At that time, only a few effective AEMs (e.g. phenobarbital and bromide) were available. In the 1940s and 1950s, when anti-epileptic medications such as phenytoin became available, use of the KD declined rapidly. Nevertheless, there was an occasional resurgence of interest in the KD. In 1971, the medium-chain-triglyceride diet was devised in an attempt to provide more flexibility with respect to carbohydrates, enhanced ketosis and better palatability (Huttenlocher et al., 1971). Despite a flurry of new anti-epileptic medications in the 1990s, frustration with the medical intractability of childhood epilepsies continued and, recently, public awareness of the KD has increased. The KD is now considered a safe and effective alternative therapy for children and adult humans with refractory epilepsy, with an efficacy that rivals (and perhaps exceeds) the newer AEMs (Stafstrom, 2004a). More recently, alternative diets for epilepsy treatment, such as the Atkins diet and a lowglycaemic index diet, have been devised that represent modifications of the original KD (Stafstrom, 2004b).
One preclinical study exists attempting to induce ketosis in dogs using diet (Puchowicz et al., 2000). The study evaluated whether a diet containing one-half of the daily caloric requirement (DCR) as R,S-butanediol diacetoacetate could achieve a stable level of ketosis in the 1 to 3 mM range. This range is considered optimal for the treatment of intractable epilepsy in people on the classic KD (Ross et al., 1985; Schwartz et al., 1989; Swink et al., 1997). Because the yield of conversion of R,S-butanediol diacetoacetate to ketone bodies is 100%, and because this conversion is catalysed by processes that are not inhibited by carbohydrates, the study hypothesized that this ester could replace part or all of the long-chain triacylglycerols in the traditional KD. If successful, it would also allow for more carbohydrates in the modified KD and make this diet more acceptable for children.
However, dogs seldom become markedly ketotic (Puchowicz et al., 2000), because there is good evidence that dogs have a high capacity to utilize ketone bodies (de Bruijne and van den Brom, 1986; Balasse and Fery, 1989; Des Rosiers et al., 1990; Ciraolo et al., 1995).
Dogs were given repeated doses of R,Sbutanediol diacetoacetate at 2% of the DCR per hour (equivalent to 48% of the DCR per 24 h). At the end of the experiment, total ketone body concentration was 0.77 mM. This level of ketosis would probably be qualified as insufficient for the treatment of epileptic children. Also of concern was the rapid decrease in total ketone body concentration seen during the hours following ingestion (Puchowicz et al., 2000).
Mechanism of action
Various hypotheses have been put forth over the years to explain KD efficacy, including acidosis, cellular and extracellular dehydration, a direct action of ketone bodies (acetoacetate, b-hydroxybutyrate, acetone) on neuronal firing or synaptic function, effects of lipids on neuronal excitability, changes in the source or utilization of energy within the brain, and alterations of mitochondrial metabolism. None of these is a sufficient explanation for the effectiveness of the KD. The challenge has been to understand how an anticonvulsant effect results from a metabolic shift from carbohydrate to fat as an energy source (Freeman et al., 2006). There are multiple sites within relevant biochemical pathways where seizure suppression could be facilitated, and the mechanism by which the KD exerts an anticonvulsant effect likely involves the combination of altered energy homeostasis and regulation of neuronal and synaptic excitability. It is also conceivable that the KD may work in different ways against different seizure types and epileptic syndromes. Both clinical studies and animal experiments have been used to decipher KD mechanisms (Stafstrom, 1999). Any explanation of mechanism of action must take into account clinical observations and known biochemical alterations resulting from ingestion of the KD.
A reasonable hypothesis is that ketone bodies exercise anti-seizure activity, much like AEMs. Serum ketone concentrations rise markedly in subjects on the diet and urinary ketones are used clinically to indicate ketosis. However, several clinical and experimental studies have shown that the relationship between serum or urinary ketones and seizure control is imprecise (Huttenlocher, 1976; Bough et al., 2000b; Gilbert et al., 2000; Likhodii et al., 2000). Direct effects of ketones on excitatory and/or inhibitory neurotransmission and on ictal activity in vitro have been disappointingly negative (Stafstrom et al., 1999; Thio et al., 2000). However, in vivo, rats on the KD or a calorie-restricted normal diet exhibited increased paired-pulse inhibition in the hippocampal dentate gyrus and increased resistance to maximal dentate activation (a form of seizure activity) (Bough et al., 2000a). Acetone could also exert an anticonvulsant effect (Likhodii et al., 2000, 2003; Likhodii and Burnham, 2002; Musa-Veloso et al., 2002, 2006; Rho et al., 2002). In summary, there appears to be a ‘threshold level’ of ketosis that must be achieved and maintained for the KD to be maximally effective.
The KD may enhance the function of g-amino butyric acid (GABA), the main inhibitory neurotransmitter of the mammalian brain. GABA is synthesized from glutamate via the action of glutamate decarboxylase. In pathways of ketone body metabolism (GABA shunt), glial conversion of glutamate to glutamine (a GABA precursor) is increased by ketosis (Erecinska et al., 1996; Yudkoff et al., 2001a). Enhanced GABA accumulation or function at the receptor level could lead to reduced cortical excitability. At the same time, the rate of glutamate conversion to aspartate, which has an excitatory role, is reduced (Yudkoff et al., 2001b). In ketosis, a-ketoglutarate, the precursor of glutamate, is increased, thereby enhancing the synthesis of GABA via the ‘GABA shunt’ (Erecinska et al., 1996). Ketones mimic GABA structurally, so they could play a direct inhibitory role on cellular excitability by stimulating GABA receptors or enhancing their action. KD treatment (and calorie restriction) increases the expression of glutamic acid decarboxylase (GAD65 and GAD67) isoforms, providing a mechanism by which GABA synthesis might be increased on the KD (Cheng et al., 2004). In a study utilizing a variety of seizure induction methods, the most consistent protection with the KD was found when seizures were induced by blocking GABA receptors (i.e. picrotoxin, bicuculline).
Efficacy
The clinical efficacy of the KD in humans has been well documented through the decades. Blinded studies are difficult to perform because of the nature of the therapy, and no blinded studies of efficacy have been done. Even prior to the resurgence of interest in the KD over a decade ago, a large number of mostly retrospective studies demonstrated its efficacy. In multiple studies performed over many decades, and despite the availability of many new AEMs, the KD appears to provide excellent seizure control (>90% seizure reduction) in 20–30% of patients and improved seizure control (>50% seizure reduction) in 60–80% of patients when evaluated at 6 months. The KD does not appear to work in 20–40% of patients. Efficacy ultimately depends on a number of factors, including types of seizures, seizure aetiology, patient age, compliance with the diet, length of follow-up and methods used to determine efficacy. A prospective multi-centre study of the classical KD, using an intention-to-treat design, showed that 29% of the 51 children who had started the diet had >90% improvement at 6 months and 53% had >50% improvement (Vining et al., 1998). In a larger, similarly designed single-centre study, 32% of the 150 children who had started the diet had >90% improvement at 6 months and 51% had >50% improvement (Freeman et al., 1998; Vining et al., 1998).
In dogs, a single study exists in the literature evaluating whether a high fat, low carbohydrate food (ketogenic food; KF) had an effect on seizure frequency with idiopathic epilepsy compared to a control food (CF) (Patterson et al., 2005). A multi-institutional, prospective, double masked, placebo-controlled study was performed from 1999 to 2002. Dogs were enrolled if they had a diagnosis of idiopathic epilepsy, were receiving phenobarbital and/ or potassium bromide at steady-state blood concentrations and had at least three seizures in the previous 3 months. Over a 3- to 6-month baseline monitoring period during which all dogs were fed CF, an initial seizure frequency was established. After the baseline monitoring, dogs with five or more seizures were randomized to begin receiving CF (16% crude fat, 54% NFE, 25% crude protein; as dry matter) or KF (57% fat, 5.8% NFE, 28% protein; as dry matter) following a 36 h fast. Seizure frequency and laboratory results were evaluated at 0, 0.5, 3 and 6 months into the test period. Thirty-one dogs entered the baseline monitoring period, 17 dogs underwent randomization for the test period with 12 completing the study (6 CF, 6 KF).
Dogs in the KF group had significantly higher serum concentrations of betahydroxybutyrate (BHB) at 3 and 6 months during the test period (2.10, 1.99 mg/dl) than the CF group (0.87, 0.62 mg/dl) (p = 0.012, p = 0.0072). One-third of dogs in each group had a 50% or greater reduction in seizure frequency. There was no difference in seizure frequency between KF group dogs (2.02, 2.41/ month) and CF group dogs (2.35, 1.36/month) at 0 and 6 months respectively (p = 0.71, 0.17) despite the differences in BHB concentrations. Power calculations performed following completion of the test period indicated that 22 dogs in each group would be required to show significant differences between food groups using seizure frequency as the major outcome variable. Therefore there was insufficient power in this study to determine effect of food treatment.
Side Effects
With humans, it is difficult to determine the frequency of side-effects in children treated with the KD, as few long-term prospective studies have been undertaken. Many of the problems are mild and can be managed with conservative measures. The most common side-effects are gastrointestinal in nature, including poor appetite, nausea, vomiting and especially constipation (Kessler et al., 2011). Sometimes these problems are associated with excessive ketosis and require alterations in the diet. Constipation is usually managed by increasing fluid intake, maximizing fibre and using a laxative such as polyethylene glycol. Occasionally an enema is necessary. Renal stones are relatively common and occur in 5–7% of children on the diet (Furth et al., 2000).
These children are typically managed with increased fluids, but sometimes require either lithotripsy or surgical removal. Children with a family history of renal stones should be carefully watched, and occasional screening for haematuria appears appropriate in all patients. There is the possibility that stones can be avoided by monitoring the urine calcium: creatinine ratio. If this ratio is elevated, urine can be alkalinized. A wide variety of other problems have been reported in association with KD use, including sepsis, pancreatitis, hepatitis, cardiomyopathy, prolonged Q-T interval, optic neuropathy, anaemia, mineral and vitamin deficiencies, hypocarnitinaemia and osteopaenia (Ballaban-Gil et al., 1998; Kang et al., 2004; Kossoff, 2004).
In the canine study described above, three of nine dogs fed KF developed pancreatitis, and 2 of 31 dogs fed CF developed pancreatitis. There was no statistical difference in the incidence of pancreatitis between the two groups (Chi-square 4.61, p = 0.203). These data suggest that this population of dogs appear at risk for developing pancreatitis regardless of food fed. High owner satisfaction with the results of the diet to which their dog was assigned may have been due to close monitoring, anti-epileptic drug dose modifications, placebo-type effects and/or stable dietary chloride helping stabilize bromide levels.
Acupuncture
In traditional Chinese medicine (TCM), seizures are referred to as internal Wind and acupuncture points (acupoints) on the head and other parts of the body, known to eliminate Wind, are used. The most common acupuncture techniques used to treat seizures include needles with no electricity (dry needle acupuncture), aqua-acupuncture using B12 and implantation of gold beads or other substances into acupoints.
Acupoints are small areas, usually a few millimetres in size and located just beneath the surface of the skin. Acupoints can be located with an ohm-meter, as they have low electrical resistance compared to the surrounding areas. Anatomic structures around acupoints include free and specialized nerve endings, small arterioles, veins, connective tissue and tissue mast cells. Although all of these structures may play a role in the mechanism of action of acupuncture, the nervous system plays a predominant role. Activation of specific brain regions, unique to the particular acupoint, can be observed on magnetic resonance imaging (fMRI) during acupoint stimulation. Acupoints are located at motor points (nerve endings within muscles), along superficial nerves and their branches, at bony foramen and fascial regions where nerves enter and exit. Acupoint stimulation can alter the physiological functions and blood supply of local and distant tissues via spinal cord reflexes involving sensory afferents and somatic and visceral efferents, as well as ascending and descending pathways in the spinal cord and brain.
There are approximately 361 acupoints that have been transposed from humans and horses to dogs. Related transpositional acupoints follow lines referred to as Channels (meridians) located along fascial and muscle planes near the surface of the body. Twelve paired Channels correspond to each of the 12 TCM organ systems and contain different numbers of acupoints. These acupoints are named according to their Channel and number
(e.g. Kidney 3 or KID-3 is the third acupoint on the Kidney Channel). Two other unpaired Channels, the Governing Vessel on the dorsal midline and the Conception Vessel on the ventral midline, also contain acupoints commonly used for disease treatment and prevention.
There are 77 classical acupoints in dogs that are not associated with a specific Channel, but have Chinese names, specific locations and actions; they are often used during acupuncture treatment to achieve certain effects. For example, the classical acupoint Da-feng-men (Great Wind Gate) is located on the midline level with the cranial edge of the ear bases and is used to treat seizures.
The Channels, containing the acupoints used for treatment, represent only the exterior portion of an intricate web of connecting Channel branches that communicate with all organs, tissues and cells. Since the 1970s, studies utilizing acoustic recordings, scintigraphy, electrical recordings, thermography, various tissue specific dyes, confocal scanning and electron microscopy, computerized tomography and magnetic resonance imaging have provided insights into the structure and function of the Channels and their matrix of collateral branches (Jing Luo system) in humans and animals. During acupuncture treatment, acupoints may be stimulated with needles alone, needles with electrical stimulation, needles with heat or heat alone (moxibustion), low-level impulse lasers, injections of substances like vitamin B12 or implantation of substances (gold beads).
Mechanism of action
Acupuncture involves complex theories of regulation of the five elements (fire, earth, metal, water and wood), yin and yang, Qi, and blood and body fluids. By stimulating various meridian points, disharmony and dysregulation of organ systems is corrected to relieve symptoms and restore natural internal homeostasis (Cheuk and Wong, 2008). Many studies in animals and humans have demonstrated that acupuncture can cause multiple biological responses. These responses can occur both locally or close to the site of application and at a distance, mediated mainly by sensory neurons to many structures within the central nervous system. The result is activation of pathways affecting various physiological systems in the brain as well as in the periphery (Cheuk and Wong, 2008).
There are both anecdotal reports and animal studies that suggest acupuncture may inhibit seizures. In an experiment of penicillin-induced epilepsy in rats, electro-acupuncture (EA) was found to inhibit seizures, possibly through decreasing neuronal and inducible nitric oxide synthase transcription in the hippocampus (Yang et al., 2000). Antagonism of GABAA (gamma-aminobutyric acid) receptors was found to attenuate the anti-epileptic effect of EA, whilst electro-acupuncture acted synergistically with the antagonists of non-Nmethyl-D-aspartate (non-NMDA) receptors (Cheuk and Wong, 2008). EA may theoretically have an effect on epilepsy by increasing the release of inhibitory neurotransmitters, such as serotonin, GABA, or opioid peptides (Cheuk and Wong, 2008).
Efficacy
The therapeutic effect of acupuncture on epilepsies was evaluated in four experimental (mice and cats) models in one paper (Chen and Huang, 1984). Twenty-four acupuncture points were used. During a total of 74 seizure events the ictal activity was suppressed in four and aggravated in 66.
A 75% or greater reduction in seizure frequency after dry needle acupuncture was reported in two human clinical randomized controlled trials (Chen and Huang, 1984), comparing acupuncture and phenytoin. Three other human trials comparing cat gut implantation at acupoints and valproate, reported a 75% or greater reduction in seizure frequency in the acupoint implantation groups (Cheuk and Wong, 2008). Reviewers of human clinical trials have concluded that larger and better-designed studies were needed before acupuncture recommendations for human epilepsy could be made.
Five epileptic dogs, nonresponsive to high levels of anticonvulsants, were presented to the acupuncture clinic at the Veterinary Hospital of the University of Pennsylvania, USA for treatment (Klide et al., 1987). Acupuncture was performed by placing small gold implants subcutaneously over the calvaria at acupuncture points on the Governing Vessel (GV), Gall Bladder (GB) and Bladder (B) meridians and left in place to provide constant stimulation to the points. Each of the five dogs treated showed a change in seizure patterns following gold implant placement. Two dogs had decreases in seizure frequency with their medication continued as before acupuncture, but they reverted to their previous pattern approximately 5 months after treatment. Three dogs continued to have decreased numbers of seizures and were maintained on decreased levels of anticonvulsants.
Another study evaluated the effect of gold wire implants in acupuncture points in dogs with uncontrolled idiopathic epileptic seizures (Goiz-Marquez et al., 2009). Fifteen dogs with such diagnosis were enrolled in the study. An EEG recording was performed in all dogs under anaesthesia with xylazine (1 mg/kg) and propofol (6 mg/kg) before the treatment protocol, and a second EEG was performed 15 weeks later. Relative frequency power, intrahemispheric coherence available through EEG, number of seizures and seizure severity were compared before and after treatment. There were no significant statistical differences before and after treatment in relative power or in intrahemispheric coherence in the EEG recording. However, there was a significant mean difference in seizure frequency and seizure severity between control and treatment periods. After treatment, nine of the 15 dogs (60%) had at least a 50% reduction in seizure frequency during the 15 weeks established as follow-up of this treatment.
Side effects
Seizures may be induced by EA in some dogs; therefore, although used in laboratory animal models evaluating seizures, EA is generally avoided in the clinical treatment of seizures in dogs and cats. Gold bead implantation at acupoints on the head may result in artifacts that can interfere with interpretation of future MRI and CT brain scans.
Surgical Therapy
The goal of any treatment for epilepsy is to permit the patient to live as normal a life as possible (Martlé et al., 2013). Maximizing normal function and minimizing adverse effects is part of the overall goal of therapy, whether medical or surgical. Two broad categories of surgical therapy for epilepsy, curative and palliative, define the relative success of surgical intervention. Curative surgery eradicates seizures and the need for medication, whereas palliative surgery lessens seizure severity or frequency or prevents the occurrence of some seizure types.
Whenever surgery for epilepsy is contemplated, the risks and benefits must be carefully weighed. The risks of epilepsy surgery are acceptably low in people in the modern era, with overall mortality being less than 0.5% and morbidity 5%. Surgical complications include cerebral infarction, intracranial haemorrhage, intracranial infection and direct cranial nerve or cerebral injury, possibly resulting in temporary or permanent neurologic deficits. Morbidity and mortality vary according to the patient’s age and type of surgery; risks appear to be slightly higher in children compared with adults, and in hemispherectomy and corpus callosotomy compared to anterior temporal lobectomy and extratemporal resections. The risks of surgery must also be compared with the risks of continued medical treatment. At present, few data address the relative risks of medical versus surgical treatment. Risks associated with ongoing epilepsy and their medical treatments include death, injury, SE, possible detrimental effects of seizures and adverse effects of medication. People with epilepsy have increased mortality rates compared with the general population (Leestma et al., 1989), and a preliminary report suggests that this risk might be reduced by epilepsy surgery (Ventureyra and Higgins, 1993). A large prospective controlled study of patients with persistent seizures revealed that achieving complete seizure control after epilepsy surgery reduces mortality to a level that is indistinguishable from the general population, whereas seizure persistence continues to be associated with high mortality rates (Sperling et al., 1999). Thus, given the reduced risk of death after successful epilepsy surgery, the long-term risks of medical therapy exceed the risks of epilepsy surgery in suitable candidates.
Surgical procedures in humans to treat epilepsy include lesionectomy, lobectomy, corticectomy, multiple subpial transection, corpus callosotomy and various combinations of these procedures. For hemispheric epilepsy syndromes, various forms of hemispheric resection and disconnection are utilized. The specific type of surgery employed for a patient depends on the predominant seizure type, the location of the seizure focus, the presence of a demonstrable lesion and the patient’s cognitive and neurologic status. The timing of surgery must take into account the natural history of the epilepsy syndrome, the patient’s developmental status and, in children, issues related to cerebral plasticity.
Disorders of the mesial temporal lobe
Disorders of the mesial temporal lobe in humans often give rise to seizures that ultimately become refractory to medical treatment, constituting the most common surgically remediable epilepsy syndrome. Although mesial temporal lobe epilepsy (MTLE) can be caused by tumours, vascular malformations, developmental anomalies and other discrete epileptogenic lesions that are potentially resectable, within this condition the most common syndrome is MTLE with hippocampal sclerosis (French et al., 1993; Williamson et al., 1993). There is often a history of febrile seizures or other neurologic insults in early childhood (Harvey et al., 1995), and complex partial seizures typically begin in the first or second decade of life. The syndrome is additionally characterized by anterior or mid-temporal spikes on EEG and hippocampal atrophy and signal abnormality on MRI (Jackson et al., 1990). Additionally, patients may have temporal lobe hypometabolism on PET (Engel et al., 1982), temporal lobe hypoperfusion on single photon-emission computed tomography (SPECT) (Rowe et al., 1991) and specific memory disturbances. Thus all patients with medically intractable TLE should be given consideration for surgery, early in the course of their epilepsy. For patients with unilateral or predominantly unilateral seizures, most centres perform a standard anterior temporal lobectomy in which the amygdala, anterior hippocampus and anterior temporal neocortex are resected (Falconer et al., 1955). Intraoperative electrocorticography and cortical stimulation are used at some centres to tailor the lateral temporal resection according to the extent of EEG abnormality and the location of the language cortex (Duchowny et al., 1992). Selective amygdalo-hippocampectomy, sparing the lateral temporal neocortex, is performed at some centres (Yasargil et al., 1985), while other centres perform only lateral neocortical resections, sparing amygdala and hippocampus. The aim of modified temporal resections is to reduce post-operative cognitive deficits; however, the indications for these procedures are not standardized and, as a result, surgical decisions are largely based on local institutional philosophy and the results of preoperative MRI and neuropsychological testing.
Lesional neocortical epilepsy
Lesional neocortical epilepsy is a focal epilepsy associated with a discrete neocortical lesion such as a tumour, vascular malformation or focal cortical dysplasia and constitutes another surgically remediable epilepsy syndrome (Cascino et al., 1993; Palmini et al., 1994). In this setting, the potential exists to cure the patient’s seizures with minimal electrophysiologic investigation and localized resection. However, surrounding cortex may harbour occult pathology and be epileptogenic, necessitating more thorough electrophysiologic investigation and wider resection (Berger et al., 1991, 1993). For lesions that do not involve critical cortex, the surgeon can perform a generous resection that includes the lesion and surrounding cortex. When lesions are close to or directly involve language, cognitive, motor or sensory cortex, excisional surgery may be contraindicated or may only be feasible with stereotactically guided lesionectomy (Cascino et al., 1990), corticectomy or lesionectomy guided by EEG localization and functional mapping (Berger et al., 1993), or resection combined with multiple subpial transection (Morrell et al., 1989). Patients with large lesions, such as large areas of pachygyria, may still benefit from epilepsy surgery but only by extensive resection of the lesion and surrounding epileptogenic cortex. For humans with multiple lesions, successful surgery is still possible if seizures can be localized to a single lesion or group of lesions (Bebin et al., 1993).
‘Non-lesional’ neocortical epilepsy
‘Non-lesional’ neocortical epilepsy represents a challenging subgroup of human cases, which could benefit from surgery. Many have extratemporal epileptogenic regions and present with a wide spectrum of simple partial, complex focal, or secondarily generalized seizure types. When the seizure semiology suggests a well-localized site of onset, the scalp EEG shows localized discharges, and the patient’s neurologic examination and cognitive profile do not suggest extensive cerebral dysfunction, surgery is a reasonable consideration. By contrast, surgery is often unrewarding in patients with poorly localized electroclinical findings or evidence of pervasive cerebral dysfunction.
Hemispheric epilepsy syndromes
Hemispherectomy is generally indicated in patients with widespread unilateral EEG abnormalities, diffuse unilateral structural abnormality and clinical evidence of hemiparesis and hemianopia. In young patients, hemispherectomy may be performed in the absence of all these features in hopes of greater seizure-freedom and more complete transfer of function to the other hemisphere. Detailed pre-operative EEG monitoring and functional neuroimaging are usually unnecessary to lateralize seizures but may help to exclude contralateral seizure onset and confirm the functional integrity of the contralateral hemisphere. Various surgical techniques are employed to treat hemispheric epilepsy syndromes, and these include anatomic hemispherectomy, functional hemispherectomy, hemispherectomy and hemi-decortication. The choice of technique depends in part on the patient’s age, the type of lesion, the size of the hemisphere and lateral ventricle and the surgeon’s expertise.
Surgical therapy in veterinary patients
The surgical procedures described above are possible to some extent in dogs and the pros and cons have been documented in the literature (Bagley et al., 1996). One study exists evaluating the effects of longitudinal division of the corpus callosum in dogs as a potential surgery for epilepsy treatment (Bagley et al., 1995). The surgery was performed in six normal beagles to determine surgical morbidity. The corpus callosum was divided sagittally on the midline and the effect on neurological function was determined. Five of six dogs were clinically normal within 14 days or less after surgery. One dog had persistent but improving clinical signs consistent with a forebrain disturbance at 30 days after surgery. Since this time, no further work has been done in this area and no clinical epileptic dogs have been evaluated.
Herbal Medicine
Herbal medicines have been used to treat seizures for centuries. Arguably the earliest use of herbal medicines for epilepsy dates back to 6000 bc in India. According to old Chinese medical texts, herbal epilepsy treatments started in China about 3000 bc. In Peru, iconographies on stones dating back to about 3000 bc describe the medicinal use of Saint Peter herb (Trichocereus peruvianis). Persian medieval medical texts document the clinical manifestations and herbal treatments of epilepsy. Numerous substances were used to treat seizures over the past 500 years in Europe and in pre-Columbian America. In 1763, Pedro de Horta in Mexico wrote an extensive monograph on epilepsy, including the use of herbal medicines, earning his title as the first American epileptologist. Today, many herbal medicines around the world are anecdotally believed to have anticonvulsant properties in humans, generally with inadequate scientific support.
Traditional Chinese medicine
The use of herbal medicines in China has a long and sophisticated tradition based on a well-developed system of medical principles. Today, traditional Chinese herbal medicine is widely practised. Similarly, in Japan, over 100 herbal (Kampo) medicines are reimbursed by the national health system and widely prescribed.
Herbal medicines traditionally used to treat convulsive diseases in Asia include: ChaiHu-Long-Ku-Mu-Li-Tan, a mixture of extracts from 13 herbal medicines; Gastrodia elata; Uncaria rhynchophylla; Menispermum dauricum; Shitei-To, a mixture of extracts from three medicinal herbs, Shitei (Kaki Calyx; calyx of Diospyros kaki L. f.), Shokyo (Zin-giberis Rhizoma; rhizome of Zingiber officinale Roscoe) and Choji (Caryophylli flos; flower bud of Syzygium aromaticum [L.]); a mixture of radish and pepper (which contains the alkaloid piperine); Qingyangshen; Kanbaku-taiso-to, a mixture of three herbal drugs, Glycyrrhizae Radix, Tritici Semen and Zizyphi Fructus; Paeoniae Radix; and Zheng Tai Instant Powder (a complex prescription of TCM used for tonicclonic seizures) (Lu et al., 1994; D’Hooge et al., 1996; Guo and Kuang, 1996; Chen et al., 2000; Hu et al., 2000; Wu et al., 2000; Kim et al., 2003; Lee et al., 2003).
Published reports suggest that several of these herbs have neuroprotective properties, and hippocampal slice models, and effects on gene expression (D’Hooge et al., 1996; Chiou et al., 1997; Sugaya et al., 1997, 1999; Hsieh et al., 1999, 2000; Chen et al., 2000; Hu et al., 2000; Minami et al., 2000; Kim et al., 2001, 2003; Lee et al., 2003). Interpretation of these studies is limited, however, by inconsistent descriptions of herbal production and extraction methods as well as the lack of characterization of active ingredients.
Based on the TCM principles of holism and differentiation, practitioners recommend individualized prescriptions (formulas/ combinations) of herbal medicines and acupuncture to their patients with epilepsy. Differentiation is the process in TCM whereby different treatable syndromes are diagnosed according to various theories and principles based on symptoms, physical signs, disease history and other information gathered from other diagnostic methods. As a result, herbal formulas with varying components and acupuncture using different points are commonly recommended to different patients who may have the same seizure type, leading to methodologic issues in conducting clinical research.
In a systematic review of human clinical randomized clinical trials (RCT) evaluating seizure control, the Chinese herbal medicine Xia Xing Ci and phenytoin were equally effective in two studies and another study found Dian Xian Ning and valproate to be equally effective for different types of epilepsy (Li et al., 2009). Three other studies reported a 50% or greater reduction in seizure frequency with other Chinese herbal medicines (Li et al., 2009). After a review of the human clinical RCT of acupuncture and epilepsy, it was concluded that larger numbers of patients were needed to ensure the effectiveness and safety of Chinese herbal medicines for treating epilepsy in humans (Li et al., 2009). Veterinary clinical RCT are warranted and may scientifically support the clinical impressions of the positive effects of Chinese herbal medicines for the management of seizure disorders of dogs and cats.
Commonly used herbs
The top selling herbs in the USA include ginkgo, St John’s wort, ginseng, garlic, echinacea, saw palmetto, kava, Pycnogenol (Horphag Research, Geneva, Switzerland), cranberry, valerian root, evening primrose, bilberry and milk thistle. Herbs described as effective or possibly effective for the treatment of seizures are described below.
American Hellebore
American hellebore (Veratrum viride) has been used for diverse indications, including neuralgia, peritonitis, pneumonia and seizures. Synonyms for American hellebore are false hellebore, green hellebore, Indian poke, itchweed and swamp hellebore. The common trade name is cryptenamine. The plant is found in the USA. It contains ester alkaloids that are chemically similar to steroids. American hellebore has variable cardiovascular effects. High doses have been found to elevate blood pressure, but other resources suggest its use to lower blood pressure, heart rate and respiratory rate (Pearl et al., 2011). The plant causes cardiac tissue, nerve membranes and muscle tissue to become more highly depolarized, leading to increased muscle tone. This is a highly toxic drug with a low therapeutic index. Adverse effects on the central nervous system (CNS), heart, gastrointestinal (GI) system, lungs and autonomic nervous system have been reported in patients using American hellebore. Paresthesia, extraocular muscle paralysis, hypertonia, weakness and seizures may occur with CNS toxicity (Pearl et al., 2011). Cardiac effects include syncope and bradyarrhythmias. GI symptoms include abdominal pain and distention, nausea and vomiting. Respiratory symptoms are dyspnea and respiratory depression. Autonomic effects are increased salivation and either hypertension or hypotension.
Betony
There are many anecdotal indications for the use of betony (Stachys officinalis), including anxiety, asthma, bronchitis, diarrhoea, headache, heartburn, palpitations, renal disease, roundworm, seizures, stomach aches, toothaches and wounds. It is traditionally used as a tea although some herbalists advocate smoking betony for the treatment of headache. Despite multiple claims, there is little available evidence to support the use of betony for any therapeutic application (Pearl et al., 2011). Synonyms for betony are bishopswort and wood betony. Betony is a member of the mint family indigenous to Europe, northern Africa and Siberia. The actions of betony are thought to come from tannins, which constitute 15% of betony. Reported side effects include GI irritation (diarrhoea, nausea and anorexia), hypo-tension and hepatic dysfunction. Uterine contractions have also been reported in people (Pearl et al., 2011).
Blue Cohosh
Blue cohosh (Caulophyllum thalictroides) has been proposed for seizure reduction. The active agent, methylcytosine, has similarities to nicotine. Synonyms for blue cohosh are blue ginseng, caulophyllum, papoose toot, squawroot and yellow ginseng (Pearl et al., 2011). This plant is a perennial found in the Midwestern and Eastern USA and Canada. As suggested by the name, its seeds are bright blue. Actions in animal studies include the stimulation of smooth muscle in coronary vessels, the small intestine and the uterus (Pearl et al., 2011). Adverse reactions include chest pain, hypertension, abdominal cramps, diarrhoea, hyperglycaemia and poisoning in children after the ingestion of seeds.
Kava
Kava (Piper methysticum) is commonly used as a ceremonial beverage in the South Pacific, but its use has spread as a widely used anxiolytic. Limited studies have suggested several mechanisms of potential neuromodulatory effects. Kava is a strong L-type Ca channel inhibitor and weak Na channel blocker. Kava increases early K outward current and g-aminobutyric acid transmission. A serotonin 1A agonist effect has been described (Pittler and Ernst, 2000; Grunze et al., 2001). A meta-analysis of seven double-blind, randomized, placebo-controlled trials found some superiority over placebo for the treatment of anxiety but statistical significance in only three (Pittler and Ernst, 2000). One relatively large study of 101 patients with anxiety showed improvement (P <0.0001) on the Hamilton Anxiety Scale but not on the Clinical Global Impression Scale (Volz and Kieser, 1997). Case reports suggest usefulness in epilepsy and psychosis. Anecdotal reports claim positive results for its use to treat asthma, depression, insomnia, muscle spasms, pain, rheumatism, sexually transmitted disease and wound healing! Hyporeflexia, sedation, ataxia, headache, dizziness, vision changes, hypertension, diarrhoea, thrombocytopenia, lymphopenia, weight loss, dyspnoea, skin hypersensitivity, dopamine antagonism, conjunctival injection and haematuria have been reported by its users. The German government has investigated whether kava should be more closely regulated after reports of hepatotoxicity (Connor et al., 2001).
Mistletoe
Mistletoe (Viscum sp.) has widespread use as a remedy for various ailments, even though it has known toxic effects. In mice, mistletoe protects against pentylenetrazole-induced and bicuculline-induced seizures. No change was seen in the N-methyl-D-aspartate (tonic) seizure model (Amabeoku et al., 1998). Mistletoe has been anecdotally used to treat arteriosclerosis, cancer, depression, epilepsy, hypertension, headaches, insomnia, nervousness, sterility, tachycardia, tensions, ulcers and urinary disorders.
A number of components in mistletoe may have adverse effects, including amines, acetylcholine, choline, histamine, tyramine, flavonoids, lectins, alkaloids and acids. Human patients using mistletoe have experienced side effects, including coma, sedation, seizures, bradycardia, cardiac arrest, cardiac depression, hypotension, hypertension, hepatitis, uterine stimulation, nausea, vomiting, diarrhoea, gastritis, psychosis, miosis and mydriasis (Pearl et al., 2011). Dehydration and electrolyte imbalance should be closely monitored in patients using mistletoe.
Pipsissewa
Pipsissewa (Chimaphila umbellata) is marketed for anticonvulsant, antispasmodic and diuretic effects. Safe doses and therapeutic claims have not been evaluated in clinical studies. Synonyms are ground holly, prince’s pine, spotted wintergreen and wintergreen. The plant is a creeping perennial herb native to Eurasia and northern North America. Hypoglycaemic action in animals has been reported, as has stimulation of renal tubular function (Pearl et al., 2011). Reported side effects in humans include nausea, vomiting, diarrhoea and rash.
Skullcap
There is little clinical research regarding the use of skullcap (Scutellaria laterifolia and
S. baicalensis), and insufficient evidence recommends it for any condition or disease. Anti-inflammatory action is reportedly produced by inhibiting interleukin-1, prostaglandin E2 and leukotriene B4 (Chung et al., 1995). It may also have antioxidant effects (Shao et al., 1999). Synonyms for skullcap are helmet flower and hoodwort. The source is leaves and roots of
S. laterifolia and S. baicalensis, which are native to temperate regions of North America. Skullcap is marketed as an anticonvulsant, sedative, antihelminthic and antibacterial and is also said to have anti-inflammatory and cholesterol-lowering effects. The use of skullcap may result in hepatotoxicity, confusion, seizures, stupor, cardiac arrhythmias and fasciculations. Liver function studies should be monitored while using skullcap. Commercial forms are often adulterated with other herbs and alcohol.
Valerian
Valerian (Valeriana officinalis) is a commonly used sleep aid. Synonyms for valerian are all-heal, baldrian, cat’s love and wild valerian. Valerian has been used as an anti-epileptic for centuries and at times was considered a first-line treatment for epilepsy (Pearl et al., 2011). The chemical composition of valerian varies greatly depending where it is grown and age because decomposition occurs with time. Up to 1% of the root may be converted to isovalerate, which is structurally similar to valpropate. Valerian itself may not be well tolerated, with side effects including drowsiness and an unpleasant taste and odour analogous to sweaty socks. Valerian has a historic record of importance in epilepsy, but there is no confirmatory evidence of anti-epileptic effects (Spinella, 2001).
Melatonin
Melatonin is marketed as a dietary supplement, and its widespread use warrants discussion. Melatonin is a chronobiotic, a term analogous to nutribiotic, which is applied to vitamins and herbs. Melatonin is a derivative of serotonin metabolism and is normally produced by the pineal gland and secreted to the hypothalamus where it promotes sleep. Some have reported an anti-epileptic effect of melatonin, and animal models suggest a link between seizures and melatonin (Pearl et al., 2011). In animals, antimelatonin antibody can induce seizures (Fariello et al., 1977). Removing the pineal gland, where melatonin is made, produces seizures in rats (Philo and Reiter, 1978).
The oral bioavailability of melatonin is 10–15% absorption, and metabolism is via the cytochrome P450 (1A2 enzyme) system of the liver. Melatonin crosses the blood-brain barrier rapidly; the elimination half-life is 30–50 min.
A recent meta-analysis of nine randomized controlled trials with over 425 subjects showed no evidence of adverse effects with use limited to 3 months (Buscemi et al., 2006). In a study of 23 children with intractable epilepsy, 10% reported minor adverse effects, specifically headache, rash and abdominal pain, which did not mandate cessation of the drug (Elkhayat et al., 2010).
Herbal and homeopathic therapy for seizures in dogs and cats
Although dogs and cats with epilepsy are anecdotally treated with herbal and homeopathic therapies, the peer-reviewed literature contains no controlled clinical research of homeopathy in cats and very little in dogs (Mathie et al., 2010). A search using Pub Med found the following two studies but caution is urged in their interpretation.
Belladonna 200C was evaluated in ten dogs with idiopathic epilepsy (Varshney, 2007). During the seizure phase, three to four drops of Belladonna 200C were administered orally at 15 min intervals until reduction in seizure activity was noted, then repeated four times daily. Four dogs described with ‘head shaking syndrome’ in addition to seizures were given Cocculus 6C, three to four drops orally weekly for 3 months in addition. Seizure frequency reduced during the first 2 weeks post-therapy and then became ‘occasional’ in the next 2 weeks. With continuation of belladonna therapy, no seizures were observed during 2–7 months follow-up. In two dogs, seizures reappeared within 15–25 days of cessation of therapy. Belladonna therapy was resumed and seizure control was again achieved. Owners were advised to continue the therapy at least twice daily until there were no fits for 2–3 months. Liver-specific enzymes were monitored, no abnormalities were observed.
A Bernese mountain dog was diagnosed with focal seizures, confirmed by electroencephalographic findings (Schneider et al., 2009). Treatment with Huperzine A, a compound isolated from Chinese club moss with NMDA receptor blocking activity, anticholinesterase activity and anticonvulsant properties, suppressed the abnormal behavior for several months but the effect was not long standing.
Side effects
Both underreporting of herbal and supplement products by human patients and animal
owners alike and the lack of recognition of potential herb–drug interactions represent important issues in clinical practice. There are three major types of interactions involving herbs and epilepsy: pharmacokinetic effects, the effects of herbs on drug metabolism; pharmacodynamic effects, herb–drug interactions that occur in the brain and other organ systems but are not predictable based on pharmacokinetic principles of absorption and metabolism; and direct effects of herbs on the seizure threshold. Two principal pharmacologic systems are affected by herbs: the P450 and P-glycoprotein systems.
The P450 hepatic enzyme system may be induced or inhibited by medications including herbs. Several commonly used herbs interact with the P450 system (Table 25.1). This potentially results in unexpected (and unrecognized) subtherapeutic anti-epileptic medication levels or toxicity.
Of the many transport systems, Pglycoprotein (Pgp) is the most thoroughly studied. Pgp is an adenosine triphosphatedependent pump that extrudes substrates out of cells. Pgp is controlled by the multidrugresistance gene MDR-1. Pgp contributes to the blood-brain barrier and limits the entry of drugs into the brain via the choroid plexus and cerebral endothelium. Pgp is also found in the intestinal epithelium, where it limits absorption from the gut. MDR-1 expression, and therefore Pgp transport, is affected by many naturally occurring compounds (Cott, 2001). Herbs that affect Pgp are St John’s wort, garlic, pycnogenol and pipsissewa.
Some herbs used for the treatment of epilepsy may interfere with the actions of drugs used for the treatment of other conditions.
Table 25.1. Effects of commonly used herbs on drug metabolism.
Herb Effect on P450 activity1
Garlic Inhibits Echinacea Inhibits Pycnogel Inhibits Milk thistle Inhibits American hellebore Inhibits Mugwort Inhibits
1Based on literature
Betony, for example, has been shown to lower blood pressure and may exacerbate the effects of antihypertensive medications. Blue cohosh has hypoglycaemic properties, thereby aggravating antidiabetic medications. Alcohol, benzodiazepines and barbiturates may increase the toxicity of kava (Almeida and Grimsley, 1996). The impurity of some herbs may further lead to unpredictable results.
Some herbs used to treat seizures, such as American hellebore, mistletoe and skullcap, may actually exacerbate seizures (Table 25.2). Ginkgo, used to enhance cognition and prevent mental decline with ageing, has been found to cause seizures in some individuals. Certain herbal oils (e.g. eucalyptus, fennel, hyssop, pennyroyal, rosemary, sage, savin, tansy, thuja, turpentine and wormwood) used for aromatherapy, cooking and cleaning have been reported to cause seizures. These observations are based on oral consumption and transdermal absorption. Although not an herbal drug, the ubiquitous compound caffeine has been associated with the aggravation of seizures (Kaufman and Sachdeo, 2003; Bonilha and Li, 2004). Additionally, it has been used in the past to prolong seizures in patients receiving electroshock therapy. There is evidence that acute and chronic caffeine administration impairs the anticonvulsant activity of some antiseizure medications in the murine maximal electroshock model (Chroscinska-Krawczyk et al., 2009).
Table 25.2. Effects of commonly used herbs on seizure activity.
Herb Effect on seizure activity1
Gingko Proconvulsant Evening primrose Proconvulsant Ephedra Proconvulsant Mistletoe Anticonvulsant and
proconvulsant American hellebore Anticonvulsant and proconvulsant Skullcap Anticonvulsant and proconvulsant
1Based on literature
References
Almeida, J.C. and Grimsley, E.W. (1996) Coma from the health food store: interaction between kava and alprazolam. Annals of Internal Medicine 125, 940–941.
Amabeoku, G.J., Leng, M.J. and Syce, J.A. (1998) Antimicrobial and anticonvulsant activities of Viscum capense. Journal of Ethnopharmacology 61, 237–241.
Arrol, L., Penderis, J., Garosi, L., Cripps, P., Gutierrez-Quintana, R. and Goncalves, R. (2012) Aetiology and long-term outcome of juvenile epilepsy in 136 dogs. The Veterinary Record 170, 335.
Asconape, J.J., Moore, D.D., Zipes, D.P., Hartman, L.M. and Duffell, W.H., Jr (1999) Bradycardia and asystole with the use of vagus nerve stimulation for the treatment of epilepsy: a rare complication of intraoperative device testing. Epilepsia 40, 1452–1454.
Bagley, R.S., Baszler, T.V., Harrington, M.L., Pluhar, G.E., Moore, M.P., Keegan, R.D. and Greene, S.A. (1995) Clinical effects of longitudinal division of the corpus callosum in normal dogs. Veterinary Surgery 24, 122–127.
Bagley, R.S., Harrington, M.L. and Moore, M.P. (1996) Surgical treatments for seizure. Adaptability for dogs. Veterinary Clinics of North America - Small Animal Practice 26, 827–842.
Balasse, E.O. and Fery, F. (1989) Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes/Metabolism Reviews 5, 247–270.
Ballaban-Gil, K., Callahan, C., O’Dell, C., Pappo, M., Moshe, S. and Shinnar, S. (1998) Complications of the ketogenic diet. Epilepsia 39, 744–748.
Bebin, E.M., Kelly, P.J. and Gomez, M.R. (1993) Surgical treatment for epilepsy in cerebral tuberous sclerosis. Epilepsia 34, 651–657.
Ben-Menachem, E., Manon-Espaillat, R., Ristanovic, R., Wilder, B.J., Stefan, H., Mirza, W., Tarver, W.B. and Wernicke, J.F. (1994) Vagus nerve stimulation for treatment of partial seizures: 1. A controlled study of effect on seizures. First International Vagus Nerve Stimulation Study Group. Epilepsia 35, 616–626.
Berendt, M., Gredal, H., Pedersen, L.G., Alban, L. and Alving, J. (2002) A cross-sectional study of epilepsy in Danish Labrador Retrievers: prevalence and selected risk factors. Journal of Veterinary Internal Medicine/ American College of Veterinary Internal Medicine 16, 262–268.
Berendt, M., Gredal, H., Ersbøll, A.K. and Alving, J. (2007) Premature death, risk factors, and life patterns in dogs with epilepsy. Journal of Veterinary Internal Medicine/American College of Veterinary Internal Medicine 21, 754–759.
Berger, M.S., Ghatan, S., Geyer, J.R., Keles, G.E. and Ojemann, G.A. (1991) Seizure outcome in children with hemispheric tumors and associated intractable epilepsy: the role of tumor removal combined with seizure foci resection. Pediatric Neurosurgery 17, 185–191.
Berger, M.S., Ghatan, S., Haglund, M.M., Dobbins, J. and Ojemann, G.A. (1993) Low-grade gliomas associated with intractable epilepsy: seizure outcome utilizing electrocorticography during tumor resection. Journal of Neurosurgery 79, 62–69.
Bonilha, L. and Li, L.M. (2004) Heavy coffee drinking and epilepsy. Seizure 13, 284–285.
Boon, P., Vonck, K., Van Walleghem, P., D’Have, M., Goossens, L., Vandekerckhove, T., Caemaert, J. and De Reuck, J. (2001) Programmed and magnet-induced vagus nerve stimulation for refractory epilepsy. Journal of Clinical Neurophysiology 18, 402–407.
Bough, K.J., Matthews, P.J. and Eagles, D.A. (2000a) A ketogenic diet has different effects upon seizures induced by maximal electroshock and by pentylenetetrazole infusion. Epilepsy Research 38, 105–114.
Bough, K.J., Yao, S.G. and Eagles, D.A. (2000b) Higher ketogenic diet ratios confer protection from seizures without neurotoxicity. Epilepsy Research 38, 15–25.
Buscemi, N., Vandermeer, B., Hooton, N., Pandya, R., Tjosvold, L., Hartling, L., Vohra, S., Klassen, T.P. and Baker, G. (2006) Efficacy and safety of exogenous melatonin for secondary sleep disorders and sleep disorders accompanying sleep restriction: meta-analysis. British Medical Journal 332, 385–393.
Cantello, R., Rossi, S., Varrasi, C., Ulivelli, M., Civardi, C., Bartalini, S., Vatti, G., Cincotta, M., Borgheresi, A., Zaccara, G., Quartarone, A., Crupi, D., Lagana, A., Inghilleri, M., Giallonardo, A.T., Berardelli, A., Pacifici, L., Ferreri, F., Tombini, M., Gilio, F., Quarato, P., Conte, A., Manganotti, P., Bongiovanni, L.G., Monaco, F., Ferrante, D. and Rossini, P.M. (2007) Slow repetitive TMS for drug-resistant epilepsy: clinical and EEG findings of a placebo-controlled trial. Epilepsia 48, 366–374.
Cascino, G.D., Kelly, P.J., Hirschorn, K.A., Marsh, W.R. and Sharbrough, F.W. (1990) Stereotactic resection of intra-axial cerebral lesions in partial epilepsy. Mayo Clinic Proceedings 65, 1053–1060.
Cascino, G.D., Andermann, F., Berkovic, S.F., Kuzniecky, R.I., Sharbrough, F.W., Keene, D.L., Bladin, P.F., Kelly, P.J., Olivier, A. and Feindel, W. (1993) Gelastic seizures and hypothalamic hamartomas: evaluation of patients undergoing chronic intracranial EEG monitoring and outcome of surgical treatment. Neurology 43, 747–750.
Chachua, T., Bilanishvili, I., Khizanishvili, N. and Nanobashvili, Z. (2010) Noradrenergic modulation of seizure activity. Georgian Medical News 34–39.
Chase, M.H. and Nakamura, Y. (1968) EEG response to afferent abdominal vagal stimulation. Electroencephalography and Clinical Neurophysiology 24, 396.
Chase, M.H., Nakamura, Y., Clemente, C.D. and Sterman, M.B. (1967) Afferent vagal stimulation: neurographic correlates of induced EEG synchronization and desynchronization. Brain Research 5, 236–249.
Chen, L.C., Chou, M.H., Lin, M.F. and Yang, L.L. (2000) Lack of pharmacokinetic interaction between valproic acid and a traditional Chinese medicine, Paeoniae Radix, in healthy volunteers. Journal of Clinical Pharmacy and Therapeutics 25, 453–459.
Chen, R.C. and Huang, Y.H. (1984) Acupuncture on experimental epilepsies. Proceedings of the National Science Council, Repubic of China B 8, 72–77.
Cheng, C.M., Hicks, K., Wang, J., Eagles, D.A. and Bondy, C.A. (2004) Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. Journal of Neuroscience Research 77, 270–276.
Cheuk, D.K. and Wong, V. (2008) Acupuncture for epilepsy. Cochrane Database Syst Rev, CD005062.
Chiou, L.C., Ling, J.Y. and Chang, C.C. (1997) Chinese herb constituent beta-eudesmol alleviated the electroshock seizures in mice and electrographic seizures in rat hippocampal slices. Neuroscience Letters 231, 171–174.
Chroscinska-Krawczyk, M., Ratnaraj, N., Patsalos, P.N. and Czuczwar, S.J. (2009) Effect of caffeine on the anticonvulsant effects of oxcarbazepine, lamotrigine and tiagabine in a mouse model of generalized tonic-clonic seizures. Pharmacology Reports 61, 819–826.
Chung, C.P., Park, J.B. and Bae, K.H. (1995) Pharmacological effects of methanolic extract from the root of Scutellaria baicalensis and its flavonoids on human gingival fibroblast. Planta Medica 61, 150–153.
Ciraolo, S.T., Previs, S.F., Fernandez, C.A., Agarwal, K.C., David, F., Koshy, J., Lucas, D., Tammaro, A., Stevens, M.P., Tserng, K.Y. et al. (1995) Model of extreme hypoglycemia in dogs made ketotic with (R,S)-1,3-butanediol acetoacetate esters. American Journal of Physiology 269, E67–75.
Connor, K.M., Davidson, J.R. and Churchill, L.E. (2001) Adverse-effect profile of kava. CNS Spectrums 6, 848, 850–853.
Cooper, I.S., Amin, I. and Gilman, S. (1973) The effect of chronic cerebellar stimulation upon epilepsy in man. Transactions of the American Neurological Association 98, 192–196.
Cooper, I.S., Upton, A.R. and Amin, I. (1980) Reversibility of chronic neurologic deficits. Some effects of electrical stimulation of the thalamus and internal capsule in man. Applied Neurophysiology 43, 244–258.
Cott, J.M. (2001) Herb-drug interactions: focus on pharmacokinetics. CNS Spectrum 6, 827–832.
D’Hooge, R., Pei, Y.Q., Raes, A., Lebrun, P., Van Bogaert, P.P. and De Deyn, P.P. (1996) Anticonvulsant activity of piperine on seizures induced by excitatory amino acid receptor agonists. Arzneimittelforschung 46, 557–560.
Daniele, O., Brighina, F., Piazza, A., Giglia, G., Scalia, S. and Fierro, B. (2003) Low-frequency transcranial magnetic stimulation in patients with cortical dysplasia – a preliminary study. Journal of Neurology 250, 761–762.
De Bruijne, J.J. and van den Brom, W.E. (1986) The effect of long-term fasting on ketone body metabolism in the dog. Comparative Biochemistry and Physiology B 83, 391–395.
DeGiorgio, C.M., Schachter, S.C., Handforth, A., Salinsky, M., Thompson, J., Uthman, B., Reed, R., Collins, S., Tecoma, E., Morris, G.L., Vaughn, B., Naritoku, D.K., Henry, T., Labar, D., Gilmartin, R., Labiner, D., Osorio, I., Ristanovic, R., Jones, J., Murphy, J., Ney, G., Wheless, J., Lewis, P. and Heck, C. (2000) Prospective long-term study of vagus nerve stimulation for the treatment of refractory seizures. Epilepsia 41, 1195–1200.
Deng, Z.D., Peterchev, A.V. and Lisanby, S.H. (2008) Coil design considerations for deep-brain transcranial magnetic stimulation (dTMS). Conference Proceedings IEEE Engineering in Medicine and Biology Society 2008, 5675–5679.
Des Rosiers, C., Montgomery, J.A., Garneau, M., David, F., Mamer, O.A., Daloze, P., Toffolo, G., Cobelli, C., Landau, B.R. and Brunengraber, H. (1990) Pseudoketogenesis in hepatectomized dogs. American Journal of Physiology 258, E519–528.
Dietrich, S., Smith, J., Scherzinger, C., Hofmann-Preiss, K., Freitag, T., Eisenkolb, A. and Ringler, R. (2008) [A novel transcutaneous vagus nerve stimulation leads to brainstem and cerebral activations measured by functional MRI]. Biomed Tech (Berl) 53, 104–111.
Duchowny, M., Levin, B., Jayakar, P., Resnick, T., Alvarez, L., Morrison, G. and Dean, P. (1992) Temporal lobectomy in early childhood. Epilepsia 33, 298–303.
Ebus, S.C., Majoie, H.J., Arends, J.B. and Boon, P.J. (2004) Can spikes predict seizure frequency? Results of a pilot study in severe childhood epilepsies treated with vagus nerve stimulation. Seizure 13, 494–498.
Elkhayat, H.A., Hassanein, S.M., Tomoum, H.Y., Abd-Elhamid, I.A., Asaad, T. and Elwakkad, A.S. (2010) Melatonin and sleep-related problems in children with intractable epilepsy. Pediatric Neurology 42, 249–254.
Elliott, R.E., Rodgers, S.D., Bassani, L., Morsi, A., Geller, E.B., Carlson, C., Devinsky, O. and Doyle, W.K. (2011) Vagus nerve stimulation for children with treatment-resistant epilepsy: a consecutive series of 141 cases. Journal of Neurosurgery - Pediatrics 7, 491–500.
Engel, J., Jr, Brown, W.J., Kuhl, D.E., Phelps, M.E., Mazziotta, J.C. and Crandall, P.H. (1982) Pathological findings underlying focal temporal lobe hypometabolism in partial epilepsy. Annals of Neurology 12, 518–528.
Erecinska, M., Nelson, D., Daikhin, Y. and Yudkoff, M. (1996) Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. Journal of Neurochemistry 67, 2325–2334.
Falconer, M.A., Meyer, A., Hill, D., Mitchell, W. and Pond, D.A. (1955) Treatment of temporal-lobe epilepsy by temporal lobectomy; a survey of findings and results. Lancet 268, 827–835.
Fariello, R.G., Bubenik, G.A., Brown, G.M. and Grota, L.J. (1977) Epileptogenic action of intraventricularly injected antimelatonin antibody. Neurology 27, 567–570.
Finnen, M.J. and Lovell, C.R. (1991) Purification and characterisation of phospholipase A2 from human epidermis. Biochemical Society Transactions 19, 91S.
Fisher, R.S. (2012) Therapeutic devices for epilepsy. Annals of Neurology 71, 157–168.
Fisher, R.S. and Handforth, A. (1999) Reassessment: vagus nerve stimulation for epilepsy: a report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 53, 666–669.
Fisher, R.S., Uematsu, S., Krauss, G.L., Cysyk, B.J., Mcpherson, R., Lesser, R.P., Gordon, B., Schwerdt, P. and Rise, M. (1992) Placebo-controlled pilot study of centromedian thalamic stimulation in treatment of intractable seizures. Epilepsia 33, 841–851.
Fisher, R., Salanova, V., Witt, T., Worth, R., Henry, T., Gross, R., Oommen, K., Osorio, I., Nazzaro, J., Labar, D., Kaplitt, M., Sperling, M., Sandok, E., Neal, J., Handforth, A., Stern, J., Desalles, A., Chung, S., Shetter, A., Bergen, D., Bakay, R., Henderson, J., French, J., Baltuch, G., Rosenfeld, W., Youkilis, A., Marks, W., Garcia, P., Barbaro, N., Fountain, N., Bazil, C., Goodman, R., Mckhann, G., Babu Krishnamurthy, K., Papavassiliou, S., Epstein, C., Pollard, J., Tonder, L., Grebin, J., Coffey, R. and Graves, N. (2010) Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia 51, 899–908.
Fornai, F., Ruffoli, R., Giorgi, F.S. and Paparelli, A. (2011) The role of locus coeruleus in the antiepileptic activity induced by vagus nerve stimulation. European Journal of Neuroscience 33, 2169–2178.
Fraser, D.D., Hoehn, K., Weiss, S. and Macvicar, B.A. (1993) Arachidonic acid inhibits sodium currents and synaptic transmission in cultured striatal neurons. Neuron 11, 633–644.
Frediani, T., Lucarelli, S., Pelliccia, A., Vagnucci, B., Cerminara, C., Barbato, M. and Cardi, E. (2001) Allergy and childhood epilepsy: a close relationship? Acta Neurologica Scandanavica 104, 349–352.
Frediani, T., Pelliccia, A., Aprile, A., Ferri, E. and Lucarelli, S. (2004) Partial idiopathic epilepsy: recovery after allergen-free diet. La Pediatria Medica et Chirurgica 26, 196–197.
Freeman, J., Veggiotti, P., Lanzi, G., Tagliabue, A. and Perucca, E. (2006) The ketogenic diet: from molecular mechanisms to clinical effects. Epilepsy Research 68, 145–180.
Freeman, J.M., Vining, E.P., Pillas, D.J., Pyzik, P.L., Casey, J.C. and Kelly, L.M. (1998) The efficacy of the ketogenic diet-1998: a prospective evaluation of intervention in 150 children. Pediatrics 102, 1358–1363.
Fregni, F., Otachi, P.T., Do Valle, A., Boggio, P.S., Thut, G., Rigonatti, S.P., Pascual-Leone, A. and Valente, K.D. (2006) A randomized clinical trial of repetitive transcranial magnetic stimulation in patients with refractory epilepsy. Annals of Neurology 60, 447–455.
French, J.A., Williamson, P.D., Thadani, V.M., Darcey, T.M., Mattson, R.H., Spencer, S.S. and Spencer, D.D. (1993) Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Annals of Neurology 34, 774–780.
Furth, S.L., Casey, J.C., Pyzik, P.L., Neu, A.M., Docimo, S.G., Vining, E.P., Freeman, J.M. and Fivush, B.A. (2000) Risk factors for urolithiasis in children on the ketogenic diet. Pediatriatric Nephrology (Berlin) 15, 125–128.
George, R., Salinsky, M., Kuzniecky, R., Rosenfeld, W., Bergen, D., Tarver, W.B. and Wernicke, J.F. (1994) Vagus nerve stimulation for treatment of partial seizures: 3. Long-term follow-up on first 67 patients exiting a controlled study. First International Vagus Nerve Stimulation Study Group. Epilepsia 35, 637–643.
Gilbert, D.L., Pyzik, P.L. and Freeman, J.M. (2000) The ketogenic diet: seizure control correlates better with serum beta-hydroxybutyrate than with urine ketones. Journal of Child Neurology 15, 787–790.
Gildenberg, P.L. (2005) Evolution of neuromodulation. Stereotactic and Functional Neurosurgery 83, 71–79.
Giorgi, F.S., Pizzanelli, C., Biagioni, F., Murri, L. and Fornai, F. (2004) The role of norepinephrine in epilepsy: from the bench to the bedside. Neuroscience and Biobehavioral Reviews 28, 507–524.
Giorgi, F.S., Mauceli, G., Blandini, F., Ruggieri, S., Paparelli, A., Murri, L. and Fornai, F. (2006) Locus coeruleus and neuronal plasticity in a model of focal limbic epilepsy. Epilepsia 47(Suppl. 5), 21–25.
Goiz-Marquez, G., Caballero, S., Solis, H., Rodriguez, C. and Sumano, H. (2009) Electroencephalographic evaluation of gold wire implants inserted in acupuncture points in dogs with epileptic seizures. Research in Veterinary Science 86, 152–161.
Graber, K.D. and Fisher, R.S. (2012) Deep Brain Stimulation for Epilepsy: Animal Models. In: Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W. and Delgado-Escueta, A.V. (eds) Jasper’s Basic Mechanisms of the Epilepsies, 4th edn. National Center for Biotechnology Information, Bethesda, Maryland.
Graves, N.M. and Fisher, R.S. (2005) Neurostimulation for epilepsy, including a pilot study of anterior nucleus stimulation. Clinical Neurosurgery 52, 127–134.
Grunze, H., Langosch, J., Schirrmacher, K., Bingmann, D., Von Wegerer, J. and Walden, J. (2001) Kava pyrones exert effects on neuronal transmission and transmembraneous cation currents similar to established mood stabilizers – a review. Progress in Neuropsychopharmacology and Biological Psychiatry 25, 1555–1570.
Guo, Q. and Kuang, P. (1996) Studies of Qingyangshen (II): modulatory effect of co-treatment with qingyangshen and diphenylhydantoin sodium on rat hippocampal c-fos expression during seizures. Journal of Traditional Chinese Medicine 16, 48–50.
Haag, M. (2003) Essential fatty acids and the brain. Canadian Journal of Psychiatry 48, 195–203.
Hammond, E.J., Uthman, B.M., Reid, S.A. and Wilder, B.J. (1992) Electrophysiological studies of cervical vagus nerve stimulation in humans: I. EEG effects. Epilepsia 33, 1013–1020.
Handforth, A., Degiorgio, C.M., Schachter, S.C., Uthman, B.M., Naritoku, D.K., Tecoma, E.S., Henry, T.R., Collins, S.D., Vaughn, B.V., Gilmartin, R.C., Labar, D.R., Morris, G.L., 3rd, Salinsky, M.C., Osorio, I., Ristanovic, R.K., Labiner, D.M., Jones, J.C., Murphy, J.V., Ney, G.C. and Wheless, J.W. (1998) Vagus nerve stimulation therapy for partial-onset seizures: a randomized active-control trial. Neurology 51, 48–55.
Harvey, A.S., Grattan-Smith, J.D., Desmond, P.M., Chow, C.W. and Berkovic, S.F. (1995) Febrile seizures and hippocampal sclerosis: frequent and related findings in intractable temporal lobe epilepsy of childhood. Pediatric Neurology 12, 201–206.
Henry, T.R., Votaw, J.R., Pennell, P.B., Epstein, C.M., Bakay, R.A., Faber, T.L., Grafton, S.T. and Hoffman, J.M. (1999) Acute blood flow changes and efficacy of vagus nerve stimulation in partial epilepsy. Neurology 52, 1166–1173.
Heynold, Y., Faissler, D., Steffen, F. and Jaggy, A. (1997) Clinical, epidemiological and treatment results of idiopathic epilepsy in 54 labrador retrievers: a long-term study. Journal of Small Animal Practice 38, 7–14.
Hsieh, C.L., Tang, N.Y., Chiang, S.Y., Hsieh, C.T. and Lin, J.G. (1999) Anticonvulsive and free radical scavenging actions of two herbs, Uncaria rhynchophylla (MIQ) Jack and Gastrodia elata Bl., in kainic acid-treated rats. Life Sciences 65, 2071–2082.
Hsieh, C.L., Chang, C.H., Chiang, S.Y., Li, T.C., Tang, N.Y., Pon, C.Z., Hsieh, C.T. and Lin, J.G. (2000) Anticonvulsive and free radical scavenging activities of vanillyl alcohol in ferric chloride-induced epileptic seizures in Sprague-Dawley rats. Life Sciences 67, 1185–1195.
Hu, H., Zhou, Y., Sui, Y. and Qi, M. (2000) An experimental study of effect of zheng tai instant powder on grand mal epilepsy. Journal of Traditional Chinese Medicine 20, 210–215.
Huf, R.L., Mamelak, A. and Kneedy-Cayem, K. (2005) Vagus nerve stimulation therapy: 2-year prospective open-label study of 40 subjects with refractory epilepsy and low IQ who are living in long-term care facilities. Epilepsy and Behavior 6, 417–423.
Hulkkonen, J., Koskikallio, E., Rainesalo, S., Keranen, T., Hurme, M. and Peltola, J. (2004) The balance of inhibitory and excitatory cytokines is differently regulated in vivo and in vitro among therapy resistant epilepsy patients. Epilepsy Research 59, 199–205.
Huttenlocher, P.R. (1976) Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatric Research 10, 536–540.
Huttenlocher, P.R., Wilbourn, A.J. and Signore, J.M. (1971) Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 21, 1097–1103.
Jackson, G.D., Berkovic, S.F., Tress, B.M., Kalnins, R.M., Fabinyi, G.C. and Bladin, P.F. (1990) Hippocampal sclerosis can be reliably detected by magnetic resonance imaging. Neurology 40, 1869–1875.
Janszky, J., Hoppe, M., Behne, F., Tuxhorn, I., Pannek, H.W. and Ebner, A. (2005) Vagus nerve stimulation: predictors of seizure freedom. Journal of Neurology, Neurosurgery, and Psychiatry 76, 384–389.
Jarvinen, K.M., Turpeinen, M. and Suomalainen, H. (2003) Concurrent cereal allergy in children with cow’s milk allergy manifested with atopic dermatitis. Clinical and Experimental Allergy 33, 1060–1066.
Kang, H.C., Chung, D.E., Kim, D.W. and Kim, H.D. (2004) Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia 45, 1116–1123.
Kaufman, K.R. and Sachdeo, R.C. (2003) Caffeinated beverages and decreased seizure control. Seizure 12, 519–521.
Kessler, S.K., Neal, E.G., Camfield, C.S. and Kossoff, E.H. (2011) Dietary therapies for epilepsy: future research. Epilepsy and Behavior 22, 17–22.
Kim, H.J., Moon, K.D., Oh, S.Y., Kim, S.P. and Lee, S.R. (2001) Ether fraction of methanol extracts of Gastrodia elata, a traditional medicinal herb, protects against kainic acid-induced neuronal damage in the mouse hippocampus. Neuroscience Letters 314, 65–68.
Kim, H.J., Lee, S.R. and Moon, K.D. (2003) Ether fraction of methanol extracts of Gastrodia elata, medicinal herb protects against neuronal cell damage after transient global ischemia in gerbils. Phytotherapy Research 17, 909–912.
Klide, A.M., Farnbach, G.C. and Gallagher, S.M. (1987) Acupuncture therapy for the treatment of intractable, idiopathic epilepsy in five dogs. Acupuncture and Electrotherapeutics Research 12, 71–74.
Kossoff, E.H. (2004) More fat and fewer seizures: dietary therapies for epilepsy. Lancet Neurology 3, 415–420.
Krahl, S.E., Clark, K.B., Smith, D.C. and Browning, R.A. (1998) Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia 39, 709–714.
Krahl, S.E., Senanayake, S.S. and Handforth, A. (2003) Right-sided vagus nerve stimulation reduces generalized seizure severity in rats as effectively as left-sided. Epilepsy Research 56, 1–4.
Kratz, O., Studer, P., Barth, W., Wangler, S., Hoegl, T., Heinrich, H. and Moll, G.H. (2011) Seizure in a nonpredisposed individual induced by single-pulse transcranial magnetic stimulation. Journal ECT 27, 48–50.
Krauss, G.L. and Fisher, R.S. (1993) Cerebellar and thalamic stimulation for epilepsy. Advances in Neurology 63, 231–245.
Labar, D., Murphy, J. and Tecoma, E. (1999) Vagus nerve stimulation for medication-resistant generalized epilepsy. E04 VNS Study Group. Neurology 52, 1510–1512.
Lee, J., Son, D., Lee, P., Kim, S.Y., Kim, H., Kim, C.J. and Lim, E. (2003) Alkaloid fraction of Uncaria rhynchophylla protects against N-methyl-D-aspartate-induced apoptosis in rat hippocampal slices. Neuroscience Letters 348, 51–55.
Leestma, J.E., Walczak, T., Hughes, J.R., Kalelkar, M.B. and Teas, S.S. (1989) A prospective study on sudden unexpected death in epilepsy. Annals of Neurology 26, 195–203.
Li, Q., Chen, X., He, L. and Zhou, D. (2009) Traditional Chinese medicine for epilepsy. Cochrane Database Syst Rev, CD006454.
Likhodii, S.S. and Burnham, W.M. (2002) Ketogenic diet: does acetone stop seizures? Medical Science Monitor 8, HY19–24.
Likhodii, S.S., Musa, K., Mendonca, A., Dell, C., Burnham, W.M. and Cunnane, S.C. (2000) Dietary fat, ketosis, and seizure resistance in rats on the ketogenic diet. Epilepsia 41, 1400–1410.
Likhodii, S.S., Serbanescu, I., Cortez, M.A., Murphy, P., Snead, O.C., 3rd and Burnham, W.M. (2003) Anticonvulsant properties of acetone, a brain ketone elevated by the ketogenic diet. Annals of Neurology 54, 219–226.
Lockard, J.S., Congdon, W.C. and Ducharme, L.L. (1990) Feasibility and safety of vagal stimulation in monkey model. Epilepsia 31(Suppl. 2), S20–26.
Lockman, J. and Fisher, R.S. (2009) Therapeutic brain stimulation for epilepsy. Neurologic Clinics 27, 1031–1040.
Lu, Y.M., Frostl, W., Dreessen, J. and Knopfel, T. (1994) P-type calcium channels are blocked by the alkaloid daurisoline. Neuroreport 5, 1489–1492.
Martelli, A., De Chiara, A., Corvo, M., Restani, P. and Fiocchi, A. (2002) Beef allergy in children with cow’s milk allergy; cow’s milk allergy in children with beef allergy. Annals of Allergy, Asthma and Immunology 89, 38–43.
Martlé, V., Van Ham, L., Raedt, R., Vonck, K., Boon, P. and Bhatti, S. (2013) Non-pharmacological treatment options for refractory epilepsy: An overview of human treatment modalities and their potential utility in dogs. Veterinary Journal 30 Oct, S1090-0233(13)00483-8. doi: 10.1016/j.tvjl.2013.09.055. [Epub ahead of print]
Mathie, R.T., Baitson, E.S., Hansen, L., Elliott, M.F. and Hoare, J. (2010) Homeopathic prescribing for chronic conditions in feline and canine veterinary practice. Homeopathy 99, 243–248.
Matthews, H., Granger, N., Wood, J. and Skelly, B. (2012) Effects of essential fatty acid supplementation in dogs with idiopathic epilepsy: a clinical trial. Veterinary Journal 191, 396–368.
McGregor, A., Wheless, J., Baumgartner, J. and Bettis, D. (2005) Right-sided vagus nerve stimulation as a treatment for refractory epilepsy in humans. Epilepsia 46, 91–96.
Merton, P.A. and Morton, H.B. (1980) Stimulation of the cerebral cortex in the intact human subject. Nature 285, 227.
Michael, J.E., Wegener, K. and Barnes, D.W. (1993) Vagus nerve stimulation for intractable seizures: one year follow-up. Journal of Neuroscience Nursing 25, 362–366.
Minami, E., Shibata, H., Nomoto, M. and Fukuda, T. (2000) Effect of shitei-to, a traditional Chinese medicine formulation, on pentylenetetrazol-induced kindling in mice. Phytomedicine 7, 69–72.
Morales, O.G., Henry, M.E., Nobler, M.S., Wassermann, E.M. and Lisanby, S.H. (2005) Electroconvulsive therapy and repetitive transcranial magnetic stimulation in children and adolescents: a review and report of two cases of epilepsia partialis continua. Child and Adolescent Psychiatric Clinics of North America 14, 193–210, viii–ix.
Morrell, F., Whisler, W.W. and Bleck, T.P. (1989) Multiple subpial transection: a new approach to the surgical treatment of focal epilepsy. Journal of Neurosurgery 70, 231–239.
Morris, G.L., 3rd and Mueller, W.M. (1999) Long-term treatment with vagus nerve stimulation in patients with refractory epilepsy. The Vagus Nerve Stimulation Study Group E01-E05. Neurology 53, 1731–1735.
Muñana, K.R., Vitek, S.M., Tarver, W.B., Saito, M., Skeen, T.M., Sharp, N.J., Olby, N.J. and Haglund, M.M. (2002) Use of vagal nerve stimulation as a treatment for refractory epilepsy in dogs. Journal of the American Veterinary Medical Association 221, 977–983.
Musa-Veloso, K., Rarama, E., Comeau, F., Curtis, R. and Cunnane, S. (2002) Epilepsy and the ketogenic diet: assessment of ketosis in children using breath acetone. Pediatrics Research 52, 443–448.
Musa-Veloso, K., Likhodii, S.S., Rarama, E., Benoit, S., Liu, Y.M., Chartrand, D., Curtis, R., Carmant, L., Lortie, A., Comeau, F.J. and Cunnane, S.C. (2006) Breath acetone predicts plasma ketone bodies in children with epilepsy on a ketogenic diet. Nutrition 22, 1–8.
Naffah-Mazzacoratti, M.G., Bellissimo, M.I. and Cavalheiro, E.A. (1995) Profile of prostaglandin levels in the rat hippocampus in pilocarpine model of epilepsy. Neurochemistry International 27, 461–466.
Obata, T., Nagakura, T., Masaki, T., Maekawa, K. and Yamashita, K. (1999) Eicosapentaenoic acid inhibits prostaglandin D2 generation by inhibiting cyclo-oxygenase-2 in cultured human mast cells. Clinical and Experimental Allergy 29, 1129–1135.
Oshinsky, M.L. and Gomonchareonsiri, S. (2007) Episodic dural stimulation in awake rats: a model for recurrent headache. Headache 47, 1026–1036.
Palmini, A., Gambardella, A., Andermann, F., Dubeau, F., Da Costa, J.C., Olivier, A., Tampieri, D., Robitaille, Y., Paglioli, E., Paglioli Neto, E. et al. (1994) Operative strategies for patients with cortical dysplastic lesions and intractable epilepsy. Epilepsia 35(Suppl. 6), S57–71.
Patterson, E.E., Muñana, K.K., Kirk, C.A., Lowry, S.R. and Armstrong, P.J. (2005) Results of a ketogenic food trial for dogs with idiopathic epilepsy. Journal of Veterinary Internal Medicine 19, 421.
Pearl, P.L., Drillings, I.M. and Conry, J.A. (2011) Herbs in epilepsy: evidence for efficacy, toxicity, and interactions. Seminars in Pediatric Neurology 18, 203–208.
Pelliccia, A., Lucarelli, S., Frediani, T., D’Ambrini, G., Cerminara, C., Barbato, M., Vagnucci, B. and Cardi, E. (1999) Partial cryptogenetic epilepsy and food allergy/intolerance. A causal or a chance relationship? Reflections on three clinical cases. Minerva Pediatrica 51, 153–157.
Penry, J.K. and Dean, J.C. (1990) Prevention of intractable partial seizures by intermittent vagal stimulation in humans: preliminary results. Epilepsia 31(Suppl. 2), S40–43.
Philo, R. and Reiter, R.J. (1978) Characterization of pinealectomy induced convulsions in the Mongolian gerbil (Meriones unguiculatus). Epilepsia 19, 485–492.
Pischon, T., Hankinson, S.E., Hotamisligil, G.S., Rifai, N., Willett, W.C. and Rimm, E.B. (2003) Habitual dietary intake of n-3 and n-6 fatty acids in relation to inflammatory markers among US men and women. Circulation 108, 155–160.
Pittler, M.H. and Ernst, E. (2000) Efficacy of kava extract for treating anxiety: systematic review and metaanalysis. Journal of Clinical Psychopharmacology 20, 84–89.
Podell, M. and Fenner, W.R. (1993) Bromide therapy in refractory canine idiopathic epilepsy. Journal of Veterinary Internal Medicine/American College of Veterinary Internal Medicine 7, 318–327.
Puchowicz, M.A., Smith, C.L., Bomont, C., Koshy, J., David, F. and Brunengraber, H. (2000) Dog model of therapeutic ketosis induced by oral administration of R,S-1,3-butanediol diacetoacetate. Journal of Nutritional Biochemistry 11, 281–287.
Ramsay, R.E., Uthman, B.M., Augustinsson, L.E., Upton, A.R., Naritoku, D., Willis, J., Treig, T., Barolat, G. and Wernicke, J.F. (1994) Vagus nerve stimulation for treatment of partial seizures: 2. Safety, side effects, and tolerability. First International Vagus Nerve Stimulation Study Group. Epilepsia 35, 627–636.
Rho, J.M. and Stafstrom, C.E. (2012) The ketogenic diet: what has science taught us? Epilepsy Research 100, 210–217.
Rho, J.M., Anderson, G.D., Donevan, S.D. and White, H.S. (2002) Acetoacetate, acetone, and dibenzylamine (a contaminant in l-(+)-beta-hydroxybutyrate) exhibit direct anticonvulsant actions in vivo. Epilepsia 43, 358–361.
Roosevelt, R.W., Smith, D.C., Clough, R.W., Jensen, R.A. and Browning, R.A. (2006) Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain Research 1119, 124–132.
Ross, D.L., Swaiman, K.F., TorResearch F. and Hansen, J. (1985) Early biochemical and EEG correlates of the ketogenic diet in children with atypical absence epilepsy. Pediatric Neurology 1, 104–108.
Rossi, S., Hallett, M., Rossini, P.M. and Pascual-Leone, A. (2009) Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clinical Neurophysiology 120, 2008–2039.
Rotenberg, A., Depositario-Cabacar, D., Bae, E.H., Harini, C., Pascual-Leone, A. and Takeoka, M. (2008) Transient suppression of seizures by repetitive transcranial magnetic stimulation in a case of Rasmussen’s encephalitis. Epilepsy and Behavior 13, 260–262.
Rowe, C.C., Berkovic, S.F., Austin, M.C., Saling, M., Kalnins, R.M., Mckay, W.J. and Bladin, P.F. (1991) Visual and quantitative analysis of interictal SPECT with technetium-99m-HMPAO in temporal lobe epilepsy. Journal of Nuclear Medicine 32, 1688–1694.
Ruffoli, R., Giorgi, F.S., Pizzanelli, C., Murri, L., Paparelli, A. and Fornai, F. (2011) The chemical neuroanatomy of vagus nerve stimulation. Journal of Chemical Neuroanatomy 42, 288–296.
Salinsky, M.C. and Burchiel, K.J. (1993) Vagus nerve stimulation has no effect on awake EEG rhythms in humans. Epilepsia 34, 299–304.
Salinsky, M.C., Uthman, B.M., Ristanovic, R.K., Wernicke, J.F. and Tarver, W.B. (1996) Vagus nerve stimulation for the treatment of medically intractable seizures. Results of a 1-year open-extension trial. Vagus Nerve Stimulation Study Group. Archives of Neurology 53, 1176–1180.
Schachter, S.C. and Saper, C.B. (1998) Vagus nerve stimulation. Epilepsia 39, 677–686.
Schlanger, S., Shinitzky, M. and Yam, D. (2002) Diet enriched with omega-3 fatty acids alleviates convulsion symptoms in epilepsy patients. Epilepsia 43, 103–104.
Schneider, B.M., Dodman, N.H., Faissler, D. and Ogata, N. (2009) Clinical use of an herbal-derived compound (Huperzine A) to treat putative complex partial seizures in a dog. Epilepsy and Behavior 15, 529–534.
Schwartz, R.M., Boyes, S. and Aynsley-Green, A. (1989) Metabolic effects of three ketogenic diets in the treatment of severe epilepsy. Developmental Medicine and Child Neurology 31, 152–160.
Schwartz-Porsche, D., Loscher, W. and Frey, H.H. (1985) Therapeutic efficacy of phenobarbital and primidone in canine epilepsy: a comparison. Journal of Veterinary Pharmacology and Therapeutics 8, 113–119.
Shao, Z.H., Li, C.Q., Vanden Hoek, T.L., Becker, L.B., Schumacker, P.T., Wu, J.A., Attele, A.S. and Yuan, C.S. (1999) Extract from Scutellaria baicalensis Georgi attenuates oxidant stress in cardiomyocytes. Journal of Molecular and Cellular Cardiology 31, 1885–1895.
Sperling, M.R., Feldman, H., Kinman, J., Liporace, J.D. and O’Connor, M.J. (1999) Seizure control and mortality in epilepsy. Annals of Neurology 46, 45–50.
Spinella, M. (2001) Herbal medicines and epilepsy: the potential for benefit and adverse effects. Epilepsy and Behavior 2, 524–532.
Stafstrom, C.E. (1999) Animal models of the ketogenic diet: what have we learned, what can we learn? Epilepsy Research 37, 241–259.
Stafstrom, C.E. (2004a) Dietary approaches to epilepsy treatment: old and new options on the menu. Epilepsy Currents 4, 215–222.
Stafstrom, C.E. (2004b) Eating your way to seizure control. Epilepsy Currents 4, 135–136.
Stafstrom, C.E., Wang, C. and Jensen, F.E. (1999) Electrophysiological observations in hippocampal slices from rats treated with the ketogenic diet. Developmental Neuroscience 21, 393–399.
Sugaya, E., Sugaya, A., Kajiwara, K., Yuyama, N., Tsuda, T., Motoki, M., Shimizu-Nishikawa, K. and Kimura, M. (1997) Nervous diseases and Kampo (Japanese herbal) medicine: a new paradigm of therapy against intractable nervous diseases. Brain Development 19, 93–103.
Sugaya, E., Yuyama, N., Kajiwara, K., Tsuda, T., Ohguchi, H., Shimizu-Nishikawa, K., Kimura, M. and Sugaya, A. (1999) Regulation of gene expression by herbal medicines – a new paradigm of gene therapy for multi-focal abnormalities of genes. Research Communications in Molecular Pathology and Pharmacology 106, 171–180.
Swink, T.D., Vining, E.P. and Freeman, J.M. (1997) The ketogenic diet: 1997. Advances in Pediatrics 44, 297–329.
Szot, P., Weinshenker, D., White, S.S., Robbins, C.A., Rust, N.C., Schwartzkroin, P.A. and Palmiter, R.D. (1999) Norepinephrine-deficient mice have increased susceptibility to seizure-inducing stimuli. Journal of Neuroscience 19, 10985–10992.
Taha, A.Y., Trepanier, M.O., Ciobanu, F.A., Taha, N.M., Ahmed, M., Zeng, Q., Cheuk, W.I., Ip, B., Filo, E., Scott, B.W., Burnham, W.M. and Bazinet, R.P. (2013) A minimum of 3 months of dietary fish oil supplementation is required to raise amygdaloid afterdischarge seizure thresholds in rats – implications for treating complex partial seizures. Epilepsy and Behavior 27, 49–58.
Tatum, W.O.T., Moore, D.B., Stecker, M.M., Baltuch, G.H., French, J.A., Ferreira, J.A., Carney, P.M., Labar, D.R. and Vale, F.L. (1999) Ventricular asystole during vagus nerve stimulation for epilepsy in humans. Neurology 52, 1267–1269.
Tergau, F., Naumann, U., Paulus, W. and Steinhoff, B.J. (1999) Low-frequency repetitive transcranial magnetic stimulation improves intractable epilepsy. Lancet 353, 2209.
Theodore, W.H. and Fisher, R.S. (2004) Brain stimulation for epilepsy. Lancet Neurology 3, 111–118.
Theodore, W.H., Hunter, K., Chen, R., Vega-Bermudez, F., Boroojerdi, B., Reeves-Tyer, P., Werhahn, K., Kelley, K.R. and Cohen, L. (2002) Transcranial magnetic stimulation for the treatment of seizures: a controlled study. Neurology 59, 560–562.
Thio, L.L., Wong, M. and Yamada, K.A. (2000) Ketone bodies do not directly alter excitatory or inhibitory hippocampal synaptic transmission. Neurology 54, 325–331.
Trepanier, L.A., Van Schoick, A., Schwark, W.S. and Carrillo, J. (1998) Therapeutic serum drug concentrations in epileptic dogs treated with potassium bromide alone or in combination with other anticonvulsants: 122 cases (1992–1996). Journal of the American Veterinary Medical Association 213, 1449–1453.
Uthman, B.M., Reichl, A.M., Dean, J.C., Eisenschenk, S., Gilmore, R., Reid, S., Roper, S.N. and Wilder, B.J. (2004) Effectiveness of vagus nerve stimulation in epilepsy patients: a 12-year observation. Neurology 63, 1124–1126.
Vagus Nerve Stimulation Study Group (1995) A randomized controlled trial of chronic vagus nerve stimulation for treatment of medically intractable seizures. The Vagus Nerve Stimulation Study Group. Neurology 45, 224–230.
Varshney, J.P. (2007) Clinical management of idiopathic epilepsy in dogs with homeopathic Belladonna 200C: a case series. Homeopathy 96, 46–48.
Velasco, A.L., Velasco, F., Jimenez, F., Velasco, M., Castro, G., Carrillo-Ruiz, J.D., Fanghanel, G. and Boleaga, B. (2006) Neuromodulation of the centromedian thalamic nuclei in the treatment of generalized seizures and the improvement of the quality of life in patients with Lennox-Gastaut syndrome. Epilepsia 47, 1203–1212.
Velasco, F., Velasco, M., Ogarrio, C. and Fanghanel, G. (1987) Electrical stimulation of the centromedian thalamic nucleus in the treatment of convulsive seizures: a preliminary report. Epilepsia 28, 421–430.
Ventureyra, E.C. and Higgins, M.J. (1993) Complications of epilepsy surgery in children and adolescents. Pediatric Neurosurgery 19, 40–56.
Vezzani, A., Moneta, D., Richichi, C., Aliprandi, M., Burrows, S.J., Ravizza, T., Perego, C. and De Simoni, M.G. (2002) Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia 43(Suppl. 5), 30–35.
Vining, E.P., Freeman, J.M., Ballaban-Gil, K., Camfield, C.S., Camfield, P.R., Holmes, G.L., Shinnar, S., Shuman, R., Trevathan, E. and Wheless, J.W. (1998) A multicenter study of the efficacy of the ketogenic diet. Archives of Neurology 55, 1433–1437.
Volz, H.P. and Kieser, M. (1997) Kava-kava extract WS 1490 versus placebo in anxiety disorders – a randomized placebo-controlled 25-week outpatient trial. Pharmacopsychiatry 30, 1–5.
Vonck, K., Thadani, V., Gilbert, K., Dedeurwaerdere, S., De Groote, L., De Herdt, V., Goossens, L., Gossiaux, F., Achten, E., Thiery, E., Vingerhoets, G., Van Roost, D., Caemaert, J., De Reuck, J., Roberts, D., Williamson, P. and Boon, P. (2004) Vagus nerve stimulation for refractory epilepsy: a transatlantic experience. Journal of Clinical Neurophysiology 21, 283–289.
Voskuyl, R.A., Vreugdenhil, M., Kang, J.X. and Leaf, A. (1998) Anticonvulsant effect of polyunsaturated fatty acids in rats, using the cortical stimulation model. European Journal of Pharmacology 341, 145–152.
Wassermann, E.M. and Lisanby, S.H. (2001) Therapeutic application of repetitive transcranial magnetic stimulation: a review. Clinical Neurophysiology 112, 1367–1377.
Williamson, P.D., French, J.A., Thadani, V.M., Kim, J.H., Novelly, R.A., Spencer, S.S., Spencer, D.D. and Mattson, R.H. (1993) Characteristics of medial temporal lobe epilepsy: II. Interictal and ictal scalp electroencephalography, neuropsychological testing, neuroimaging, surgical results, and pathology. Annals of Neurology 34, 781–787.
Woodbury, D.M. and Woodbury, J.W. (1990) Effects of vagal stimulation on experimentally induced seizures in rats. Epilepsia 31(Suppl. 2), S7–19.
Wu, H.M., Huang, C.C., Li, L.H., Tsai, J.J. and Hsu, K.S. (2000) The Chinese herbal medicine Chai-Hu-LongKu-Mu-Li-Tan (TW-001) exerts anticonvulsant effects against different experimental models of seizure in rats. Japan Journal of Pharmacology 82, 247–260.
Xiao, Y. and Li, X. (1999) Polyunsaturated fatty acids modify mouse hippocampal neuronal excitability during excitotoxic or convulsant stimulation. Brain Research 846, 112–121.
Yang, A.C., Zhang, J.G., Rong, P.J., Liu, H.G., Chen, N. and Zhu, B. (2011) A new choice for the treatment of epilepsy: electrical auricula-vagus-stimulation. Medical Hypotheses 77, 244–245.
Yang, R., Huang, Z.N. and Cheng, J.S. (2000) Anticonvulsion effect of acupuncture might be related to the decrease of neuronal and inducible nitric oxide synthases. Acupuncture and Electrotherapeutics Research 25, 137–143.
Yasargil, M.G., Teddy, P.J. and Roth, P. (1985) Selective amygdalo-hippocampectomy. Operative anatomy and surgical technique. Advances and Technical Standards in Neurosurgery 12, 93–123.
Yehuda, S., Carasso, R.L. and Mostofsky, D.I. (1994) Essential fatty acid preparation (SR-3) raises the seizure threshold in rats. European Journal of Pharmacology 254, 193–198.
Yudkoff, M., Daikhin, Y., Nissim, I., Lazarow, A. and Nissim, I. (2001a) Brain amino acid metabolism and ketosis. Journal of Neuroscience Research 66, 272–281.
Yudkoff, M., Daikhin, Y., Nissim, I., Lazarow, A. and Nissim, I. (2001b) Ketogenic diet, amino acid metabolism, and seizure control. Journal of Neuroscience Research 66, 931–940.
Yuen, A.W., Sander, J.W., Fluegel, D., Patsalos, P.N., Bell, G.S., Johnson, T. and Koepp, M.J. (2005) Omega-3 fatty acid supplementation in patients with chronic epilepsy: a randomized trial. Epilepsy and Behavior 7, 253–258.
Zabara, J. (1992) Inhibition of experimental seizures in canines by repetitive vagal stimulation. Epilepsia 33, 1005–1012.
Zanchetti, A., Wang, S.C. and Moruzzi, G. (1952) The effect of vagal afferent stimulation on the EEG pattern of the cat. Electroencephalography and Clinical Neurophysiology 4, 357–361.
Glossary of Pharmacological Terminology
Luisa De Risio
Absorption refers to the process of a substance entering the blood circulation.
Area under the plasma concentration-time curve (AUC) refers to the area under the plot of plasma concentration of a drug against time after drug administration. It reflects the actual body exposure to a medication after administration of one dose and is expressed in mg*h/l. The AUC is of particular use in estimating bioavailability and total clearance of drugs. The AUC of drugs with linear kinetics is directly proportional to the administered dose and inversely proportional to the clearance of the drug.
Bioavailability (F) is the percentage of an administered dose of medication that reaches the systemic circulation. Bioavailability of a drug administered intravenously is by definition 100%. Bioavailability is less or equal to 100% for any other route of administration. Bioavailability is proportional to the total area under the plasma concentration-time curve (AUC).
Clearance (Cl) is the volume of fluid (e.g. plasma) irreversibly cleared of a medication per unit of time and represents the sum of total organ clearance. If a medication is cleared exclusively by one organ, then plasma Cl also represents clearance of that specific organ.
Distribution refers to the dispersion or dissemination of substances throughout the fluids and tissues of the body.
Elimination is the result of metabolism and excretion. With first-order elimination, the amount of drug eliminated is directly proportional to the serum drug concentration.
Elimination rate constant is a value used in pharmacokinetics to calculate the rate at which a medication is removed from the body. It is often abbreviated K or Kel. The Kel represents the fraction of drug eliminated from the body per unit of time. Kel is dependent upon clearance and the volume of distribution (Kel = Cl / Vd).
Elimination half-life (T1/2) is the time necessary for plasma, serum or blood concentrations of a medication to decrease by 50%. The half-life is determined by the metabolism and excretion rates of the medication. Elimination half-life of a medication determines the time to steady state and the amount of fluctuation in drug concentrations during a chosen dosing interval.
Enzyme induction refers to acceleration of metabolism by increase of enzyme expression.
Excretion refers to the removal of a drug from the body.
Idiosyncratic drug reactions (IDRs) are adverse drug reactions that are not related to the known pharmacological properties of the drug, develop mostly unpredictably in susceptible individuals only and do not appear to be dose related. The mechanism of IDRs is not clearly
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
Glossary of Pharmacological Terminology
understood, and although several theories have been proposed, IDRs can be generally categorized mechanistically as either immune-mediated or non-immune-mediated.
Individual therapeutic range can be defined as the range of AEM concentrations which is associated with the best achievable response in an individual, and therefore this range will differ in different individuals.
Linear pharmacokinetics refers to pharmacokinetics of a medication whose serum or plasma concentration is proportional to the administered dose and whose rate of elimination is proportional to the serum or plasma concentration.
Maximum concentration (Cmax) is the highest concentration of a medication which is achieved after dosing. This is commonly referred to as peak concentration.
Metabolism (or Biotransformation) refers to the irreversible transformation of parent compounds into metabolites.
Minimum concentration (Cmin) is the lowest concentration of a medication that occurs during a dosing interval. This is commonly referred to as trough concentration.
Non-linear pharmacokinetics refers to pharmacokinetics of a medication whose serum or plasma concentration is not proportional to the administered dose and whose rate of elimination is not proportional to the serum or plasma concentration.
Peak concentration, see Maximum concentration (Cmax).
Pharmacodynamics is the study of the biochemical and physiologic effects of drugs on the body, the mechanisms of drug action and the relationship between drug concentration and effect.
Pharmacokinetics is a discipline that uses mathematical models to describe and predict the time-course of drug concentrations in body fluids including drug absorption, distribution, metabolism and excretion.
Reference range can be defined as a range of AEM concentrations, which is quoted by a laboratory and specifies a lower limit below which a therapeutic response is relatively unlikely to occur, and an upper limit above which toxicity is relatively likely to occur. Reference ranges derive from statistical evaluation of the concentration interval at which the majority of patients showed an optimal response in a variable number of studies. Individuals may respond therapeutically below the range or adversely within the range.
Steady state is the state during which the rate of drug intake equals the rate of drug elimination.
Time to maximum concentration (Tmax) is the time required to achieve the medication maximum concentration
Time to steady state (Tss) is the time required to reach steady state and it depends on the medication’s elimination half-life, dosage (maintenance versus loading) and kinetics. Steady state is achieved in approximately 5 half-lives under conditions of first-order kinetics in a one-compartment distribution model (e.g. the medication is rapidly and evenly distributed throughout the body) and in the absence of a loading dose. The time required to reach a steady state may differ from the conventional 5 half-lives for medications metabolized by non-first-order kinetics, undergoing extensive first-pass metabolism in the liver, and/or multicompartment distribution in the body (e.g. the medication is distributed into plasma at one rate then exchanged between plasma and tissues at a different rate).
Trough concentration, see Minimum concentration (Cmin).
Volume of distribution (Vd) estimates the amount of tissue to which a medication is distributed.
Allele: This is one of a number of alternative forms of the same gene or same genetic locus (generally a group of genes). It is the alternative form of a gene for a character producing different effects. Sometimes different alleles can result in different observable phenotypic traits, such as different pigmentation. However, many variations at the genetic level result in little or no observable variation.
Alpha rhythm: Synchronized EEG activity with a frequency range of 8–13 Hz and amplitudes ranging from 20 to 60 µV, that occurs during relaxed wakefulness over the occipital region of the head when eyes are closed. Blocked by opening of the eyes.
Aura: An aura is a subjective sensation at the start of a seizure before there are observable signs. Human patients describe various sensations during their auras, including dizziness, tingling and anxiety. Common manifestations of auras in animals are hiding, seeking the owner, agitation or vomiting just before a seizure. In other cases an aura occurs alone, which constitutes a sensory seizure.
Autosomal recessive: A mode of inheritance of genetic traits located on the autosomes (the non-sex determining chromosomes). In opposition to an autosomal dominant trait, a recessive trait only becomes phenotypically apparent when two copies of a gene (two alleles) are present. In other words, the subject is homozygous for the trait. Recessive genetic disorders occur when both parents are carriers and each contributes an allele to the progeny. As both parents are heterozygous for the disorder, the chance of two disease alleles being inherited by one of their offspring is 25% (in autosomal dominant traits this is higher); 50% of the progeny (or 2/3 of the remaining ones) are carriers. When one of the parents is homozygous, the trait will only show in the offspring if the other parent is also a carrier. In that case, the chance of disease in the offspring is 50%.
Beta rhythm: EEG activity over 13 Hz (commonly around 14 Hz and 35–45 Hz) that localizes predominantly over the fronto-central regions of the head during wakefulness. Amplitude is usually below 30 µV.
Candidate gene study: The candidate gene approach to conducting genetic association studies focuses on associations between genetic variation within pre-specified genes of interest and phenotypes or disease states. This is in contrast to genome-wide association studies, which scan the entire genome for common genetic variation. Candidate genes are most often selected for study based on a priori knowledge of the gene’s biological functional impact on the trait or disease in question. The rationale behind focusing on allelic variation in specific, biologically
© L. De Risio and S. Platt 2014. Canine and Feline Epilepsy: Diagnosis and Management
(L. De Risio and S. Platt)
relevant regions of the genome is that certain mutations will directly impact the function of the gene in question, and lead to the phenotype or disease state being investigated. This approach usually uses the case-control study design to try to answer the question, ‘Is one allele of a candidate gene more frequently seen in subjects with the disease than in subjects without the disease?’ Suitable candidate genes are generally selected based on known biological, physiological, or functional relevance to the disease in question. This approach is limited by its reliance on existing knowledge about known or theoretical biology of disease.
Capacitance: The amount of electrical charge stored by a substance; denoted by C.
Complex segregation analysis (CSA): A technique within genetic epidemiology to determine whether there is evidence that a major gene underlies the distribution of a given phenotypic trait. CSA also provides evidence to whether the implicated trait is inherited in a Mendelian dominant, recessive or co-dominant manner. CSA is often a preliminary step in genetic epidemiology. The purpose of CSA is to provide initial evidence that a single gene has a major effect on a particular phenotypic trait. Only phenotypic information, not genotypic information, is required for CSA. CSA can provide evidence, but not definitively prove a trait is under the control of a single gene. Evidence from CSA studies can be used to justify which phenotypes might be appropriate for more in-depth studies such as linkage analysis. CSA requires phenotypic information on family members in a pedigree. A variety of models with different parameters and assumptions about the nature of the inheritance of the trait are fitted to the data. CSA studies may include non-genetic models, which assume the trait has no genetic component and is only determined by environmental factors, models that include environmental components as well as multi-gene heritability components, and models that include environment, multi-gene heritability and a single major gene to best fit the data.
Convulsion: A convulsion is an involuntary contraction or series of contractions of the voluntary muscles, which may be a seizure.
Delta rhythm: EEG activity under 4 Hz, usually seen during natural sleep.
Dominance: In genetics, dominance is a relationship between alleles of a single gene, in which one allele masks the phenotypic expression of another allele at the same gene locus. In the simplest case, where a gene exists in two allelic versions (designated A and B), three combinations of alleles (genotypes) are possible: AA, AB and BB. If AA and BB individuals (homozygotes) show different forms of some trait (phenotypes), and AB individuals (heterozygotes) show the same phenotype as AA individuals, then allele A is said to dominate or be dominant to or show dominance to allele B, and B is said to be recessive to A. If instead AB has the same phenotype as BB, B is said to be dominant to A. Dominance should be distinguished from epistasis, a relationship in which an allele of one gene affects the expression of an allele at a different gene.
Epilepsy: This is a chronic disorder characterized by paroxysmal brain dysfunction due to excessive neuronal discharge, and usually associated with some alteration of consciousness. The clinical manifestations of the event may vary from complex abnormalities of behaviour including generalized or focal convulsions to momentary spells of impaired consciousness. These clinical states have been subjected to a variety of classifications, none has been universally accepted to date and, accordingly, the terminologies used to describe the different types of events remain purely descriptive and non-standardized; they are variously based on: the clinical manifestations of the seizure; the pathologic substrate (i.e. hereditary, structural or cryptogenic); the location of the epileptogenic lesion (e.g. temporal); and the time of day at which the attacks occur.
Focal seizures: Any seizure due to a lesion in a specific, known area of the cerebral cortex (sometimes called partial seizures). These seizures may just affect the sensory, motor or autonomic regions of the brain causing clinical signs that reflect the involvement of these regions. Sometimes they are classified based upon whether there is a change or impairment in consciousness or not as complex or simple, respectively. Impaired consciousness is defined as the inability to respond normally to exogenous stimuli by virtue of altered awareness and/ or responsiveness.
Generalized seizures: A generalized seizure is one in which the first clinical change indicates initial involvement of both hemispheres. Consciousness may be impaired and this impairment may be the initial manifestation. Motor manifestations are bilateral. These type of seizures often consist of a loss of consciousness and generalized tonic convulsions followed by clonic convulsions. The muscles become rigid during the tonic phase of the seizure and alternately contract and relax during the clonic phase. The animal may lose bowel or bladder control, or have trouble breathing. However, they do not have to be tonic-clonic and can also be:
Genome-wide association study (GWAS): In genetic epidemiology, a genome-wide association study, also known as whole-genome association study, is an examination of many common genetic variants in different individuals to see if any variant is associated with a trait. GWAS typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like major diseases. These studies normally compare the DNA of two groups of participants: animals with the disease (cases) and similar animals without (controls). Each subject submits a sample of DNA, from which millions of genetic variants are read using SNP arrays. If one type of the variant (one allele) is more frequent in animals with the disease, the SNP is said to be ‘associated’ with the disease. The associated SNPs are then considered to mark a region of the human genome that influences the risk of disease. In contrast to methods that specifically test one or a few genetic regions, the GWAS investigate the entire genome. The approach is therefore said to be non-candidate-driven in contrast to gene-specific candidate-driven studies. GWAS identify SNPs and other variants in DNA that are associated with a disease, but cannot on their own specify which genes are causal.
Ictus: The ictus is the seizure itself. In most cases, the ictus lasts only a few minutes.
Impedance: Opposition to the passage of an alternating current through a circuit, e.g. the electrode impedance is the opposition to the current at the interface between an electrode and the adjacent tissue. When the capacitance factor is small, electrode resistance and impedance are usually numerically equal. Generally measured in kW.
K-complexes: Benign transient EEG activity seen during the early stages of quiet sleep characterized by a diphasic slow wave with large amplitude negative and positive deflections.
Locus: In genetics, a locus (plural loci) is the specific location of a gene or DNA sequence on a chromosome. A variant of the similar DNA sequence at a given locus is called an allele. The ordered list of loci known for a particular genome is called a genetic map. Gene mapping is the process of determining the locus for a particular biological trait.
LOD score method: The LOD score (logarithm (base 10) of odds) is a statistical test often used for linkage analysis in human, animal and plant populations. The LOD score compares the likelihood of obtaining the test data if the two loci are indeed linked, to the likelihood of observing the same data purely by chance. Positive LOD scores favour the presence of linkage, whereas negative LOD scores indicate that linkage is less likely. Computerized LOD score analysis is a simple way to analyse complex family pedigrees in order to determine the linkage between Mendelian traits (or between a trait and a marker, or two markers). By convention, a LOD score greater than 3.0 is considered evidence for linkage. A LOD score of +3 indicates 1000 to 1 odds that the linkage being observed did not occur by chance. On the other hand, a LOD score less than −2.0 is considered evidence to exclude linkage. Although it is very unlikely that a LOD score of 3 would be obtained from a single pedigree, the mathematical properties of the test allow data from a number of pedigrees to be combined by summing the LOD scores. It is important to keep in mind that this traditional cut-off of LOD >+3 is an arbitrary one and that the difference between certain types of linkage studies, particularly analyses of complex genetic traits with hundreds of markers, these criteria should probably be modified to a somewhat higher cut-off.
Mendelian trait: This is a characteristic/disease that is controlled by a single locus in an inheritance pattern. In such cases, a mutation in a single gene can cause a disease that is inherited according to Mendel’s laws.
Mendel’s Laws: Mendel summarized his findings in two laws: the Law of Segregation and the Law of Independent Assortment.
Multifactorial inheritance: Refers to polygenic inheritance that also includes interactions with the environment. Unlike monogenic traits, polygenic traits do not follow patterns of Mendelian inheritance (separated traits). Instead, their phenotypes typically vary along a continuous gradient.
Polygenic inheritance: Refers to inheritance of a phenotypic characteristic (trait) that is attributable to two or more genes and can be measured quantitatively.
Post-ictal period: This is the time after the seizure activity has finished. Post-ictal signs are transient clinical abnormalities in brain function that are caused by the ictus. Post-ictal signs typically last a few minutes to hours and can include confusion, blindness, ataxia and deep sleep.
Prodrome: Some patients with seizures experience a prodrome, which is long-lasting abnormality occurring hours to days before a seizure, such as restlessness or anxiety. The difference between a prodrome and an aura is that prodromes are longer lasting and not associated with abnormal electrical activity in the brain.
Resistance: Opposition to the passage of a direct current through a circuit. See Impedance. Generally measured in kW.
Seizure: A seizure (or ictus) has been defined as ‘a transient occurrence of signs due to abnormal excessive or synchronous neuronal activity in the brain’ (Fisher et al., 2005). The clinical manifestations of a seizure are sudden and transient and depend on location of onset in the brain, patterns of propagation and a variety of other factors. Seizures can affect one or more of the following functions: sensory, motor and autonomic activity, consciousness, emotional state, memory, cognition or behaviour (Fisher et al., 2005).
Sleep spindles: Benign transient EEG activity seen over the central region of the head during the early stages of quiet sleep characterized by waxing and waning groups of rhythmic waves showing frequencies of 7–14 Hz and lasting 1.5–2 s.
Theta rhythm: EEG activity occurring in the frequency range 4–7 Hz during normal active behavioural states.
Vertex sharp transients: Also known as ‘V waves’, these benign EEG transients are elicited by stimulation during drowsiness or early sleep and display highest negative amplitude over the midline vertex.
References
Chatrian, G.E., Bergamini, L., Dondey, M., Klass, D.W., Lennox-Buchthal, M. and Petersen, I. (1974) A glossary of terms most commonly used by clinical electroencephalographers. Electroencephalography and Clinical Neurophysiology 37, 538–548.
Fisher, R.S., Van Emde Boas, W., Blume, W., Elger, C., Genton, P., Lee, P. and Engel, J., Jr (2005) Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46, 470–472.
Litt, B. and Cranstoun, S. (2003) Engineering principles. In: Ebersole, J.S. and Pedley, T.A. (eds) Current Practice of Clinical Electroencephalography, 3rd edn. Lippincott Williams & Wilkins, Philadelphia, Pennsylvania, pp. 32–71.
Noachtar, S., Binnie, C., Ebersole, J.S., Mauguiere, F., Sakamoto, A. and Westmoreland, B. (1999) A glossary of terms most commonly used by clinical electroencephalographers and proposal for the report form for the EEG findings. The International Federation of Clinical Neurophysiology. Electroencephalography and Clinical Neurophysiology Supplement 52, 21–41.
This page intentionally left blank
anti-epileptic medication (AEM) (continued ) metabolism and pharmacokinetic
interactions 349, 351–352 neurological deficits 295 phenobarbital 374 pharmacokinetic parameters
healthy dogs 349, 353–354 phenobarbital (PB) 358 phenobarbitone 374, 380, 384, 390 phenytoin sodium 528 pregabalin 446 principles 526–529 reactive seizures 54, 60 SE (status epilepticus) in dogs
therapeutic monitoring 365–369 and AEM formulation 367–368 clinical use 365 guidelines 365 individual therapeutic range 365–366 and pathological states 368 and pharmacokinetic interactions 368 reference range 365–366 saliva 368–369 serum/plasma concentrations 365 time to steady state (Tss) 366–367
anti-epileptic treatment 347–370 AEM see anti-epileptic medication (AEM) in animals 348 clinician needs 347 clinico-pathological investigations 347 discontinuation 369 in humans 348 idiopathic epileptic dogs 348 long-term therapy 347 pet-owner education 369–370 reactive seizures 347 short term therapy 347
astrocytoma 180 boxers and older dogs 180 clinical signs 180 diagnosis 181 intra-axial primary CNS neoplasias 180 and oligodendroglioma 168, 183, 223 pathological findings and classification 180 prevalence 241 treatment and survival times 180 tumor location 180
bacterial CNS infections 123–130 aerobic and anaerobic organisms 123, 126 analysis, cerebrospinal fluid 126 colonization 123 CT and MRI 126 haematology 125–126
neurological signs 125 physical examination 125 polymerase chain reaction 126 prognosis 128 skull defect and subcutaneous
treatment 126, 128–130 AEMs 128 antibiotic susceptibility 126 antimicrobial medications 126, 129–130 brain abscessation 128 corticosteroids 126, 128 intravenous administration 126
barbiturates 339, 375 advantages 530 AEM 528–529 RSE 531
BBB see blood–brain barrier (BBB)
benzodiazepines (BZDs) 476–491 adverse effects 479–480, 528 AEM 526–528 clorazepate 490–491 continuous infusion 529–530 diazepam 481–485 intranasal route 509–511 lorazepam 488–490 mechanism of action 476–478 metabolism and pharmacokinetics
CYP isoenzymes 479 diffusion rate 479 in vivo affinity, drug 477 N-desmethyldiazepam (DMD) 479 neurotransmitter GABA 477 routes of administration and
single-dose effects 479 midazolam 485–488 molecular structure 476, 477 pharmacokinetic interactions 378–379,
479– 480 rectal administration 507–509 tolerance 480–481
blood-brain barrier (BBB) benzodiazepines 479 endothelial cells 33 lomustine 179 melatonin 556 multidrug transporter hypothesis 33 neuroprotection 90
brivaracetam (BRV) 468–470 adverse reactions 470 antiseizure medication 470 dosing and monitoring 470 efficacy 470 mechanism of action 468–469 metabolism and pharmacokinetics 469
Index 577
molecular structure 468 pharmacokinetic interactions 470
bromethalin 79–80 activation 79 antemortem and definitive diagnosis 79–80 clinical signs 79 exposure 79 malicious poison 79 management and prognosis 80
bromide (Br) 397–411 AEM 310 blood sampling 409 cardiac arrhythmias 408 CRI 408 diet 404–405 epileptic dogs resistant 432 GABA 398 gastrointestinal mucosa 404 hepatic dysfunction 362, 397 KBr 397 monotherapy 410 NaBr 397 neuronal receptor targets 398 oral dosages 404 pancreatitis 382 phenobarbital 405 pharmacokinetics see pharmacokinetic
interactions pseudohyperchloraemia 409 seizure control 410 serum bromide concentrations 409
BRV see brivaracetam (BRV) BZDs see benzodiazepines (BZDs)
CA see cytosine arabinoside (CA)
canine distemper (CD) 115–121 anti-epilpetic treatment 121 clinical signs 115, 120
acute 115, 120 chronic distemper encephalomyelitis 120 old dog encephalitis 120
diagnostic investigations 120–121 modified-live vaccines 121 polysystemic disease, dogs 115 prognosis 121 supportive care 121 testing, infectious disease 317 transmission 115 virus (CDV) 115
canine distemper virus (CDV) 115–121, 286, 313, 317
canine idiopathic epilepsy 207–213 autosomal recessive inheritance 212 behavioural changes 208 boxer and Shetland sheepdog 212 breed list 211 dog breeds, hereditary basis 211 epileptic dogs 208 familial transmission 208 genetic components 208, 209 and genetics 211–212 German shepherds and boxers 208 and lifespan 209–210 mix-breed dogs 208 phenobarbital and/or KBr 444 prevalence 211 recognized in breeds 208, 209 reflex seizures 208 segregation analysis 212 seizures 207, 208 standard poodles 212 tonic-clonic seizures 208
canine seizures disorders 219–231 age 228–229 breed 227–228 estimation, dog epilepsy 219 gender 228 and neurological examination 229–231
drug-naïve dogs 230–231 IE patients 229 interictal period 229 MRI and CSF analyses 231 neurobehavioural co-morbidities 229 symptomatic aetiology 231
prevalence 219–225 prognosis 230–231 reactive 225–227
intoxication 225 metabolic/toxic disorder 226–227 recurrent epileptic, dogs 225–226
sudden unexpected death in epilepsy (SUDEP) 231
cardiac arrhythmias 408 bufotoxins and bufagenins 88 myocardial ischaemia 153
cataplexy 264–266 narcolepsy see narcolepsy and sleep disorders 264–266
CD see canine distemper (CD); compulsive
disorders (CD) CDV see canine distemper virus (CDV) cEEG see continuous EEG recording (cEEG) cerebellar ataxia 136, 284–285 cerebrospinal fluid (CSF) 8, 113, 253, 313–316, 524
and aCSF 16 anaesthesia 314 cerebellomedullary cistern 314 flow 315 iatrogenic contamination 316 pathological haemorrhage 316 pleocytosis 315–316 protein concentration 316 refrigerated sample 315 seizure-associated changes 316
cerebrospinal fluid (CSF) (continued ) structural brain disease 313–316 ultrafiltrate 313 cerebrovascular accidents (CVAs) 102–112 aetiology 111, 112 clinical signs 102, 104 diagnosis 110–111 EEG 340 forebrain signs 102–110 crossbreed with peracute onset of left-sided 102, 105–106 and severe thrombocytopaenia 102, 106–107 spayed Staffordshire bull terrier with subacute progressive onset 102, 107–108 white terrier with peracute onset right-sided 102–104 haemorrhagic 102 intracranial 62 ischaemic 102 pathological process 102 post-stroke seizures and epilepsy antiepileptic treatment 110 cerebral ischaemia 109 diagnosis, dogs 109 early and late 109 epileptogenesis 109 estimation and severity 109 focal/generalized and status epilepticus 109 GABA-ergic interneurons 109 glutamate excitotoxicity 109 hypoxia, cerebral 109 ischaemic penumbra 109, 110 recovery and quality of life 112 prognosis 111–112 report, dogs and cats 102 stroke and TIA 102 treatment 111 clorazepate 477, 490–491 adverse reactions 490 CS 511 description 490 dosing and monitoring 490 efficacy 490–491 metabolism and pharmacokinetics 490 oral administration 512 tolerance 490 cluster seizures (CS) 503–513 definition 504–505 implications and risks 507–508 long-acting treatment 511–513 oral clorazepate 511 oral levetiracetam 511–512 VNS 514 mechanisms 504 periodicity 505
prevalence 505 risk factor 505–506 short-acting treatment 507–511 nasal benzodiazepines 509–511 rectal benzodiazepines 507–509 therapeutic considerations 507 CN examination see cranial nerve (CN) examination collapsing response mediator protein-2 (CRMP-2) 463–464, 466 compulsive disorders (CD) 266–270 conditioning 267 diagnosis 268–269 genotype 267 homogeneity 267–268 medical problems 267 pathophysiology 267 prognosis 270 signs 266–267 stress 267 treatment options 269–270 consciousness and behaviour levels 276–281 cat, window 276, 279 in dogs 276, 279 environment and attitude 276 forebrain diseased cat 280 hemi-neglect syndrome 280, 281 limbic system 280 obtunded and comatose animals 279 constant rate infusion (CRI) 408 intravenous 148 propofol 530 continuous EEG recording (cEEG) 325, 341, 342 corneal reflex 289 cranial nerve (CN) examination 286–291 cotton ball test 287 facial sensation 289 gag reflex 290 hearing 290 Horner’s syndrome 287 menace response 288 mild abnormalities 287 palpebral reflex 288 smell 290 strabismus and abnormal nystagmus 289 temporal and masseter muscle bulk 289 CRI see constant rate infusion (CRI) CRMP-2 see collapsing response mediator protein-2 (CRMP-2) CSF see cerebrospinal fluid (CSF) CVAs see cerebrovascular accidents (CVAs) cytochrome P450 (CYP450) 375 midazolam 477 pharmacokinetic interaction see pharmacokinetic interactions polymorphisms 213
Index 579
cytopenias 381, 455 cytosine arabinoside (CA) 148–150, 152, 175, 185
DBS see deep brain electrical stimulation (DBS) deep brain electrical stimulation (DBS) 543, 544 degenerative diseases 187–194 abiotrophy 194–195 description 187 leukodystrophies 193–194 lysosomal storage 187–190 mitochondrial encephalopathies and encephalomyelopathies 193 organic acidurias 190, 192–193 spongy 194 dexamethasone ACTH 386 anti-inflammatory doses 128 facial pruritus 383 prednisone 147 diagnostic investigation, seizures 300–318 in animals 300, 302 CSF 313–316 DTI 309 EEG 318–319 fMRI 313 functional neuroimaging 311 genetic testing 318 haematology, serum biochemistry and urinalysis 300, 302 hepatic function test 302 infectious disease testing 317–318 magnetic resonance volumetry 308–311 MRI 304–307 MRS 311–312 PET and SPECT 312–313 radiography and ultrasography 304 seizure-associated MRI changes 307–309 toxicological testing 302–304 dietary therapy 544–548 fatty acid supplementation 544–545 hypoallergenic diet 545 KD 545–548 diffusion tensor imaging (DTI) 309 DTI see diffusion tensor imaging (DTI)
EEG see electroencephalography (EEG) electroclinical syndromes 40, 325, 341 electroencephalography (EEG) 325–342 aging 336–337 alpha rhythm 334 amplitude spectrum 333 artefacts capacitance 337, 338 impedance 337
non-physiological artefacts 337 physiologic artefact 337 band power 333 beta rhythm 334 cerebral cortex 326 clinical applications 340–342 computerization and digital 325 delta band 334, 335 electrical activity 327 electrode types skin surface electrodes 329 SNEs 329 superficial electrodes 329 SWEs 329–330 waveform reproduction 328 extra- and intracellular ion currents 326 frequency power spectrum 333 instrumentation 327–328 montage 330–332 neurons and glial cells 326 neurons communication 326 pathological waveforms 338–340 burst-suppression pattern 339 epileptiform patterns 338 ictal patterns 340 paroxysm 338 sharp wave 338 spike-and-slow-wave complexes 339–341 vertex waves 339 patient handling 332–333 sleep spindles 335 software analysis 333–334 synchronicity 327, 328 theta waves 334, 335 vigilance state 335–336 visual analysis technique 334 epilepsy animal models 6 description 247–248 familial 12 paroxysmal event 248 pharmacoresistant risk factors 30–31 seizure 246– 247 voltagedependent sodium channels 2 ethylene glycol 82–84 acute renal failure 83 biotransformation 82 clinical signs 82 colorimetric spot tests 82–83 commercial antifreeze automotive product 82 diagnostic procedure 83 fomepizole and ethanol dosage 83 hypocalcaemia 82 intoxication 303 prognosis 83–84 quantitative test kit 83
ethylene glycol (continued ) toxicity 303, 304 treatment 83 exogenous toxic disorders 70–73 acute neurological signs 70–71 animals medications 71, 72 cutaneous 71 damage nervous system 70 decontamination and prevention 71 emergency treatment 71 gastrointestinal 71–73 activated charcoal 72–73 cathartics 73 colonic and gastric lavage 71–72 decontamination 71 emetics 71 infusion, IVLE 73 presumptive and definitive diagnosis 70 seizures 68– 70 urinary excreted 73
FECV see feline enteric coronavirus (FECV) felbamate 362, 443, 453–456 adverse reactions 455 description 453 dosing and monitoring 455–456 efficacy 456 mechanism of action 453–454 metabolism and pharmacokinetics 454–455 molecular structure 453, 454 pharmacokinetic interactions 455 feline enteric coronavirus (FECV) 121, 317 feline infectious peritonitis (FIP) 121–125 cats, FCov 121, 122 clinical signs 122 diagnosis 122–123 etiopathogenesis 121–122 multifocal CNS signs, domestic short hair cat 123–125 risk, pure and non-pure cats 122 treatment and prognosis 123 and viral non-FIP encephalitides 121 viruses 121 feline infectious peritonitis virus (FIPV) 121, 317 feline seizure disorders 235–242 age 238–239 breed 237–238 gender 238 neurological examination 239 prevalence 236–241 aetiological classification 236–237, 239–241 recurrent seizures 235 report, feline seizures 235 semiological classification 235–236 prognosis 240–242 survival times 240, 241
FIP see feline infectious peritonitis (FIP) fungal and algal diseases 112, 137–140
GABA see gamma-aminobutyric acid (GABA) gabapentin (GBP) 439–444 adverse reactions 443 antispastic effects 439 dosing and monitoring 443–444 efficacy 444 mechanism of action 440–441 metabolism and pharmacokinetics absorption 441 concentrations 441–442 in dogs and cats 442 dosage 442–443 oral dose 441 molecular structure 439, 440 neuropathic and post-operative pain 439 pharmacokinetic interactions 443 tolerability, safety and clinical efficacy 439 US FDA approval 439 gait examination 282–285 circling 285 motor nuclei, CN 283 non-slippery surface 282 proprioception 282 vestibular ataxia 284 gamma-aminobutyric acid (GABA) AEMs 90 antagonism 549 clorazepate 490 glutamatergic neurons 3 lorazepam 488 monoamine oxidase activity 77 oral levetiracetam 511 phenobarbital binds 374 pregabalin 444 transmembrane ion gradients 16 gamma-glutamyltransferase (GGT) hepatic enzyme 384 hyperbilirubinaemia 419 gastrointestinal mucosa 404 GBP see gabapentin (GBP) generalized tremor syndromes 258– 261 dancing doberman disease 261 episodic hypertonicity 259–260 essential/geriatric (senile) canine 259 muscle stiffness 260–261 paroxysmal dyskinesia 261 scotty cramp 259 startle disease, Irish wolfhounds 260 GGT see gamma-glutamyltransferase (GGT) glomerular filtration 400 diurnal variations 427 renal tubules 400
Index 581
glutamate 6–9, 17–18 activity 529, 547 anticonvulsant effects 4 concentrations 169, 391, 541 CVAs 108, 109 depletion 17–18 excitotoxicity 239, 464, 520 extracellular 152 and GABA 2 GLDH 86 glial buffering 18 and glutamine (Glx) 311 HE 59 hypoglycaemia 55 knockout/ knockdown procedures 6–7 receptors
AMPA 7 ionotropic and metabotropic 7, 8 NMDA 7, 33, 520 postsynaptic 15
relationship 3 release 146 reuptake 113 transporters 8 treatment 8–9
CSF 9 elevations 8 GABA concentration 9 gabapentin 9 idiopathic epilepsy, dogs 8 NMDA and AMPA receptor antagonists 8
glutamatergic and g-aminobutyric acid (GABA) 2, 6, 8–9, 18–19 GME see granulomatous meningoencephalomyelitis (GME)
granulomatous meningoencephalomyelitis (GME) characteristics, cerebrospinal fluid 114 clinical manifestation and lesion topography 145 description 145 diagnostic investigations
analysis, cerebrospinal fluid 145 haematology 145 MRI lesions 145, 145, 149–150 T2-weighted and FLAIR sequences 145
forms 145, 148 mononuclear and mixed pleocytosis 315, 316 prognosis 152 treatment
adjunctive immunosuppressive 147–148 adverse effects 147 antiepileptic 151–152 cytosine arabinoside (CA) 148, 150 corticosteroid 147 cyclosporine 150–151 ketoconazole 151 leflunomide and mycophenolate
lomustine 151 prednisolone protocol 147, 150 procarbazine 150 radiation therapy 151
white matter, brain 223 guarana (Paullinia cupana) 94– 95
hemi-neglect/inattention syndrome 280, 281, 296 hemispheric epilepsy syndromes 552 hepatic encephalopathy (HE) 58–62
abdominal ultrasound 304 acquired portosystemic shunts 58–59 biochemical disorder 58 congenital portosystemic shunts 58 diagnosis 59–60
haematological changes 59–60 hepatobiliary disease 60 liver biopsy 60 MRI and T1weighted images 60 portal vascular anomalies 60, 61
management 60–62 AEMs and levetiracetam control 60 gradual and portosystemic shunts
attenuation 62 L-carnitine 61–62 omeprazole and levetiracetam 62 protein intake and oral medications 61 reduces ammonia production 60–61 SAMe 62 seizures 62 urease negative bacteria 61
microvascular dysplasia 59 neurological and non-neurological
signs 59, 60 parenchymal disease 59, 60 pathophysiology 59 seizure disorders
aetiologies, feline 239–240 epidemology 226 urea-cycle enzyme deficiencies 59
hepatotoxicity 380–381 AEM 363 cytochrome P450 380 cytoprotective agents 384 immunosuppression 151 nodular hyperplasia 381 reactive oxygen species 380 serum biochemistry 381
herbal medicine 553–557 American hellebore (Veratrum viride) 554 Betony (Stachys officinalis) 554 Blue cohosh (Caulophyllum thalictroides) 554 description 553 and homeopathic therapy 556 Kava (Piper methysticum) 555 melatonin 556
herbal medicine (continued ) Mistletoe (Viscum sp.) 555 Pipsissewa (Chimaphila umbellata) 555 side effects 556–557 skullcap (Scutellaria laterifolia
and S. baicalensis) 555 traditional Chinese medicine 553– 554
Valerian (Valeriana officinalis) 556 hexachlorophene 84 homogeneity, CD 267–268
cognition levels 268 development 268 distraction 268
Horner’s syndrome 287, 297, 541
human epilepsy 210–214 complex inheritance 210 electroencephalography 340–342 forms 1 gene LGI1 213 genetics 211 inflammatory mechanisms 9–10 medication 213 monogenic disorders 210–211
autosomal 210 benign familial neonatal
convulsions 210–211 EEG data and MRI 210 episodic ataxia type I 211 febrile seizures type I and II 210 gene defects 210 non-Mendelian syndromes 211 pathophysiology 210
treatment acupuncture 550 bromide 397 rufinamide 471
hydrocephalus 162–167 acquired 164 bilateral divergent ventrolateral
strabismus 163, 164 cerebrospinal fluid 162 characterization 162 classification 162 congenital 162 diagnosis 164–166 dome-shaped calvaria 163 female bichon frise cross 164, 165 MRI pulse sequences 308 obstructive 184, 186 prognosis 167 treatment 166–167
hypernatraemia 64–66 causes 65 clinical and neurological signs 65 diagnostic investigations 65 oral water administration 66 plasma sodium concentration 64–65 reduction, sodium levels 65–66
hypoalbuminaemia 383 albumin synthesis 383 hepatic enzyme 384 phenobarbital 381
hypocalcaemia 66–67 calcium concentration 66 causes 66 clinical signs 66 diagnosis 66–67 diazepam 67 differential diagnoses 66 electrocardiographic monitoring 67 oral calcium and vitamin D supplementation 67 tetany and seizures 66 therapy maintenance 67
hypoglycaemia 55–56 blood glucose concentration 55 causes 55 diagnosis 55 error, cellular metabolism 190 hippo campal neurons 239 insulinoma-associated 56–58, 304 lysosomal storage diseases 190 management 55–56 minimum database abnormalities 303 movement disorders 256 neurologic signs 55 refractory 62 SE (status epilepticus) 524 seizure aetiology 47 severe brain damage, coma and death 55 syncope 261, 262
hyponatraemia 63–64 animal’s volume status 64 causes 63 clinical and neurologic signs 63–64 diagnostic investigations 64 dogs 63 hypervolemic animals 64 outcomes 63 plasma sodium concentration 63 treatment 64
idiopathic epilepsy (IE) 207–214 AEM half-life determination 367 anti-epileptic treatment 368, 369 bromide toxicity 401 canine see canine idiopathic epilepsy causes, (status epilepticus) 521 chronic and recurring seizure syndromes 207 classifications 207 definition 207 development 280, 402
Index 583
diagnosis 207 dogs 28, 219 focal seizures 220 genetics
anti-epileptic medication 213 canine epilepsy 211–212 and feline epilepsy 213–214 and human see human epilepsy
herbal and homeopathic therapy 556 ILAE 1981 and 1989 41 interictal signs 208–209 LEV (levetiracetam), cats 429 MRI 304 panniculitis 402 physical and neurological examinations 300 prevalence
feline seizures 235–242 seizure clustering 505 seizure disorders with known mutations
212– 213 signalment 274 systemic and brain abnormalities 207 thyroid function tests 385 veterinary seizures 40
idiosyncrasy 380–384, 402–403 AEMs 383–384 bronchial asthma 403 cytopenias 381 dexamethasone 383 dose reduction 402 dyskinesia 383 hepatotoxicity 380–381 hypoalbuminaemia 383 lymphadenopathy 383 N -acetylcysteine 384 pancreatitis 382–383, 402 panniculitis 402–403 pruritic dermatologic lesions 402 SAMe 384 superficial necrolytic dermatitis 381–382 thrombocytopenia 383
imepitoin (Pexion® ) 496–501 adverse reactions 498 anticonvulsant and anxiolytic properties 496, 501 clinical interactions, phenobarbital 497, 501 dosing and monitoring 498–499 efficacy 499–501 epileptic dogs administration 498 mechanism of action 496, 497 metabolite profiles 497 molecular structure 496, 497 oral administration 496–497 pharmacokinetic parameters 497, 498 repeated overdose 498
inflammatory CNS disease 112–115, 131–147 antifungal chemotherapy 141 brain neoplasia, infarction/trauma 112 cerebrospinal fluid analysis 113–114 clinical signs 112 development, intracranial pressure 114 dogs and cats 112 ehrlichial, anaplasmal, rickettsial
and mycoplasmal 112, 131–133 fungal and algal 112, 137–140 haematology 113 mix-breed dog with pseudorabies 116, 120 MRI, T2-weighted and FLAIR
images 114–115 parasitic 112, 144 peripheral blood eosinophilia 144 post-stroke seizures and epilepsy 112–113 prognosis 115 progressive cerebellar ataxia 136 protozoal 112, 134–135 secondary oedema 139, 145 treatment 115 viral, dogs and cats 112, 116–119
inflammatory mechanisms, epilepsy 10–14 ACTH 11 activation 11 brain 10–11 causes
adult rats and mice 11–12 cell loss and synaptic
reorganization 13–14 chemokines 12 double-label immunofluorescence
techniques 12–13 epileptogenic process 12 inflammation and veterinary
medicine 12, 13 mediators 13 microglial activation 12 proinflammatory cytokines 12 seizure-induced neuronal death 12
cell loss 10 CNS infections 10 glucocorticoids 11 human 9–10 IFNs and ILs 11, 13 immune and anti-inflammatory therapies 14 mediators 10 molecular targeting, drug design 14 neuronal excitability 14 pro-inflammatory agents 10 and seizures 10 TBI 11 TLE, brain 10 TNFs and TGF 11
insecticides 73–77 chlorinated hydrocarbons 76–77 organophosphates and carbamates 74–76
insecticides (continued ) diagnosis 75 exposure and toxicity 74–75 glycopyrrolate and fluid therapy 76 insect and nematode control,
dogs and cats 74–75 mechanism of action 75 parasympathetic signs 76 pralidoxime chloride (2-PAM) 76 prognosis 76
pyrethrin and pyrethroid/permethrin 73–74 Chrysanthemum cinerariaefolium 73 classification 73 clinical signs 74 diagnosis and treatment 74 etofenprox 74 exposure 74 formulations 74 mechanism of action, type I and II 74 prognosis 74
insulinoma 56–58 clinical signs 56 craniodorsal abdomen 56, 57 differential diagnosis 56–58 dogs and feeding 56 feline and hyperinsulinaemia 56 fructosamine concentration 56 histological examination 57 hypoglycaemia development 56 management 57–58 neurological signs 56 neuromuscular collapse 252 pancreatic b -islet cell neoplasia 56 pancreatic mass 56, 57 peripheral nerve disease 251 peripheral polyneuropathy 56
interictal–ictal transition mechanisms 5–6 and ictogenesis 6 nonsynaptic
active ion transport 5 changes, ionic microenvironment 5 ephaptic interaction 5–6 presynaptic terminal bursting 5
synaptic 6 International League against Epilepsy (ILAE) 28–29
involuntary movement abnormalities aetiology 257 CN examination see Cranial nerve (CN)
examination diagnostic approach 256 dyskinesia 255–256, 286 localized tremor syndromes 257–258 myoclonus 253–254, 286 myokymia and neuromyotonia 255 myotonia and tremor 286 neuromyotonia 286
paroxysmal events 253 tetanus and tetany 286 tremor 254– 255
ivermectin and macrocyclic lactones 89–91 AEMsand GABA agonist 90 autosomal recessive mutation 90 avermectins and milbemycins 89–90 clinical signs 90 CNS effects 90 diagnosis 90 intoxication 90 IVLE administration 91 neurotoxicity 390 P-gp-substrate drugs 90 prognosis 91 treatment 90
ketogenic diet (KD) 545–548 description 545–546 dogs 546 efficacy 547–548 mechanism of action 546–547 side effects 548
lacosamide (LCM) 463–468 adverse reactions 467 anti-epileptic medications 466 clinical pharmacokinetic study 466 dose toxicity studies 466–467 dosing and monitoring 467 efficacy 467–468 mechanism of action
CRMP-2 function 463–464, 466 neuronal receptor targets 463, 464 neuroprotective effects 464 sodium channels 463, 465
metabolism and pharmacokinetics 464–466 molecular structure 463, 464 population pharmacokinetic studies 466 refractory status epilepticus (RSE) 531–532
lameness 66, 220, 285 LCM see lacosamide (LCM) Lennox-Gastaut syndrome (LGS) 453, 458, 471 lesional neocortical epilepsy surgery 552 leukodystrophies 49, 193–194 LEV see levetiracetam (LEV) levamisole 91 levetiracetam (LEV) 425–434
adverse reactions 430–431 anti-epileptic medications 90, 101, 169,
348, 426 brivaracetam 468 cluster seizures (CS) 512 dosing and monitoring 431–432, 434 efficacy 432–434
Index 585
dogs 432–434 humans 432, 433 prevention, post-operative
tolerance and cost 433 exogenous toxic disorders 72 in humans 425, 433 and lacosamide 466 management, status epilepticus 530 mechanism of action 425–426, 434 metabolism and pharmacokinetics 426–430
concentrations 430 CYP450 427 extended release 427 idiopathic epilepsy, cats 427, 429 oral doses 427 parameters, dogs 427, 428 phenobarbital administration 430 plasma half-life, rats 426–427 renal elimination 427 subcutaneous and rectal administration 427
molecular structure 425, 426 oral 426, 512, 529 pharmacokinetic interactions 430–431 phenobarbitone 378, 379 post-traumatic seizures 153, 155–156 prophylactic anti-epileptics 156 refractory status epilepticus (RSE) 530 salivary therapeutic monitoring 368–369 seizures control, HE 60, 62, 425–434 tolerance 425, 434
LGS see Lennox-Gastaut syndrome (LGS) lissencephaly 15, 48, 161 localized tremor syndromes 257–258
ataxic head 258 head 257 limb tremors/myoclonus 257 non-ataxic head 258
lorazepam 488–490 dosing and monitoring 489 efficacy 489–490 interactions and adverse reactions 489 metabolism and pharmacokinetics 488–489
lysosomal storage diseases 187–190 cerebellar and forebrain dysfunction 187 clinical signs 187 diagnosis 190 metabolic products 187 mucopolysaccharidoses 187 seizures, dogs and cats 187–190
Magnetic resonance imaging (MRI), seizures 304–307 epilepsy 304 image acquisition protocols 305 interictal neurological abnormalities 305 pulse sequences, brain imaging 305, 308
signal intensity 305, 308 transverse T1W 305–307 magnetic resonance spectroscopy
(MRS) 311–312 metabolic abnormalities detection 311 and MRSI 311 and mTLE 312 seizure-associated changes 312
ma huang (Ephedra sinica) 94– 95 MDR1 see multidrug resistance 1 (MDR1) mesial temporal lobe epilepsy (MTLE) 312, 551–552 methylxanthines 91–92 metronidazole 47, 61, 89, 101, 129, 378 midazolam 477, 485–488, 529
description 485 dosing and monitoring 487 efficacy 487–488 interactions and adverse reactions 487 intranasal formulations 510, 511 metabolism and pharmacokinetics 486–487 nasal preparations 509 rectal administration 509
mimics of seizure activity electrical circuitry 244 involuntary movement abnormalities
253– 261 paroxysmal events 244, 245 phenotypic characterization 244, 246
modified Glasgow coma scale (MGCS) 153–159 MTLE see mesial temporal lobe epilepsy (MTLE) multidrug resistance 1 (MDR1) 390
autosomal recessive mutation 90 epileptic Border collies 213
narcolepsy 264–266 dogs and cats 264 hypocretin replacement therapy 265 motor tone ranging 264 pathogenesis 265 REM 265–266
nasal benzodiazepines, CS 509–511 diazepam 509 dose 509 hydroxypropyl methylcellulose
midazolam gel 510 intranasal midazolam 509 pharmacokinetic studies 509 side effects and risk 511
neoplasia 167–187 acute collapse, muscle 251 advanced imaging techniques 256 astrocytoma 180–182 cerebral 522 CNS lymphoma 183–185 insulinoma 56 intracranial 167–175
neoplasia (continued ) canine and feline 167 clinical signs 168 diagnosis 170, 174 incidence 167–168 pathologic effects 167 prevalence, cat seizures 235, 239 primary and common secondary 170–173 prognosis 175 survival 174, 175 treatment 174–175 tumour-related seizures and epilepsy 168–170 meningioma 175–180 breeds 176 canine and feline 175 clinical signs 176 diagnosis 176 pathology and classification 179–180 treatment and survival times 176–179 olfactory/frontal lobe 305 oligodendroglioma 182–183 pituitary tumours 185–187 clinical signs 185 diagnosis 185–186 secondary tumour, dogs and cats 185 size 185 treatment and survival times 186–187 primary and secondary nervous system 167 seizure aetiology 47 spinal disease 257 ultrasonography 256 neuroanatomic diagnosis, seizure 295–298 location and distribution 295 muscle disorders 298 neurological signs 296–297 neuromuscular junction 298 neuromuscular system disorders 296, 298 peripheral nerve dysfunction 298 neurochemical mechanisms, epilepsy 6–9 catecholamines 9 expression, glutamate receptor/ transporter 6–9 GABA 6 opioid peptides 9 neurological examination, seizure AEM-associated deficits 295 consciousness and behaviour levels 276, 279–280 description 276 form 275, 277–278 muscle mass and tone 293 palpation 295 spinal nerve reflexes 293–295
neuromuscular collapse 248–253 activity-associated weakness 248 decreased muscle tone 248–249 differential diagnosis 250, 251 exercise testing 249 focal versus diffuse 249 peripheral nerve, muscle/neuromuscular junction 249–250 specific diagnostic tests 250, 252–253 upper/lower motor neuron 249 neuromyopathy 402 neurostimulation 537–544 thalamic stimulation 543–544 transcranial magnetic brain stimulation 541–543 vagus nerve stimulation (VNS) 537–541 nociception evaluation 275, 295–297 non-invasive VNS 541 non-lesional neocortical epilepsy 552
obsessive compulsive disorder (OCD) see compulsive disorders (CD) organic acidurias 47, 187, 190, 192–193, 253
pancreatitis 382–383 hypertriglyceridaemia 382 insulinoma 56 KBr 382 lipase immunoreactivity 382, 403 panniculitis 402–403 erythema nodosum 403 lethargy and pyrexia 402 parasitic diseases 112, 142 paroxysmal dyskinesias 259, 261 pathophysiology, seizure activity 1–19 AEMs 1 cell population 5 cellular discharges 1 disorders, neuronal migration 15 epileptogenesis 4 excitability 4–5 individual neurons 4 neuronal microenvironment 4–5 human 1 ictogenesis 4 kindling procedures 4 limitation 15–16 local network neurons 17–19 amplification 17 burst firing properties 17 GABA-ergic inhibition 18–19 glial buffering glutamate 18 glutamate depletion 17–18 intra- and extracellular environments 18 transmission 17
mechanisms 5–14 inflammatory see inflammatory mechanisms, epilepsy interictal-ictal transition 5–6 neurochemical see neurochemical mechanisms, epilepsy nerve cell function, electrical basis 1–2 single feature 1 single neurons activation 16–17 depolarizations 16 energy failure 17 intracellular ion-activated potassium currents 16 termination 16 transmembrane ion gradients 16–17 synaptic transmission 2–3 synchronization 3–4 phenobarbital see phenobarbital (PB) penitrem A and roquefortine 87–88 periventricular heterotopia 15 pet-owner education 369–370 PGB see pregabalin (PGB) pharmacokinetic interactions 374–380, 398–404 AEM metabolism 368 AGP 378 benzodiazepines 378–379, 479 chronic administration 378 CSF 398 CYP450 inhibitors 379 diet and urine 378 furosemide 402 gastrointestinal 401 gastrointestinal tract 380 glomerular filtration 400 griseofulvin 379 halothane anaesthesia 401 hepatic microsomal enzymes 375 idiosyncrasy 402–403 intrarectal (IR) 400 intravenous and oral administration 375–377, 398, 399 levetiracetam 379 maximal plasma concentration 375 neuromyopathy 402 oxygen therapy and bronchodilators 404 phenobarbital 401 pseudohyperchloraemia 404 pulmonary lesions and pneumothorax 403 renal excretion 400 serum phenobarbital concentrations 378 thyroid function tests 404 topiramate 460 zonisamide 379 pharmacoresistant epilepsy 29–34 animals’ welfare 34 clinical risk factors 30–31 definition 29–30
AED efficacy 29–30 human medicine 29 ILAE level 1 and 2 29 phenobarbitone 29 genetic risk factors 29 mechanisms 31–34 AED therapy 31–32 drug resistance 31 drug-target hypothesis 32 multidrug transporter hypothesis 32–33 neuronal network properties 33–34 pharmacodynamics/kinetic properties 31 pseudoresistance 31 rat and dog 209 risk factors 30–31 clinical 30–31 genetic 30 seizure free and quality of life 34 phenobarbital (PB) 374–392 ACTH 386 AEMs 374 ALT 384, 385 barbiturates 528 Blood–brain barrier (BBB) 390 blood sampling 388 bromide 389 cholestasis 385 CSF 391 GABA 374 GGT 384–385 P -glycoprotein function 390 hypercholesterolaemia 385 idiosyncrasy 380–384 intracranial tumours 380 levetiracetam (LEV) 512, 530 liver function testing 302 Luminal® 389 MDR1 gene 390 mini-loading dose 387 monotherapy 389 neuronal receptor targets 374, 375 oral dosage 386 pharmacokinetic interactions see pharmacokinetic interactions plasma aldosterone concentrations 386 post-synaptic neuron 374 Quality of life 390 salivary therapeutic monitoring 368–369 serum phenobarbital concentrations 387–388 thyroid function tests 385–386 triglyceride concentrations 388 Phenoleptil® 389 pleocytosis 315–316 CSF 113 mononuclear 193 neutrophilic 111
pleurothotonus 280–282, 285 PLR see pupillary light reflex (PLR) poisonous plants 85–87
blue-green algae/cyanobacteria causes 86 clinical signs 86 diagnosis 86 growth 86 management and prognosis 87 mechanism of action 86
causes 85– 86 list 85, 86 treatments 86
postural reactions 290–293 extensor postural thrust reaction 293 hemistanding and hemiwalking 292, 293 hopping reaction 291–292 paw replacement reaction 291 proprioceptive receptors 290 visual placing 291 wheel-barrowing reaction 292
posture, animal 280–282 decerebrate rigidity 282, 284 head tilt 281, 282 head turn 280 low head carriage 281, 283 neuromuscular weakness 281 pleurothotonus 280–282 spontaneous knuckling, toes 282, 283
potassium bromide (KBr) 382 pancreatitis 382 panniculitis 402 phenobarbitone 29
pregabalin (PGB) 444–447 adverse reactions 446 dosing and monitoring 446–447 efficacy 447 mechanism of action 440, 445 metabolism and pharmacokinetics 4
45– 446 molecular structure 440, 444–445 pharmacokinetics interactions 446 tolerability, safety and clinical
efficacy 439 US FDA approval 445 prevalence of canine seizures 219–225
aetiological classification 222–225 cryptogenic epilepsy 222 head trauma 224–225 idiopathic epilepsy 221–222 inflammation 222–223 intracranial neoplasia 223–224 recurrent 222 vascular disease 225
semiological classification 219–221 aetiology and associated outcome 220, 221
autonomic and behavioural signs 219 diagnosis, focal seizures 220 focal-onset 219 generalization 220–221 IE investigation 220 pre-ictal signs 220 structural epilepsy 220 tonic-clonic seizures 219
protozoal diseases 112, 134–135
pseudohyperchloraemia 404 bromide administration 404 chloride levels 409 serum bromide concentration 409
pupillary light reflex (PLR) 114, 287–288
rapid eye movement (REM) sleep disorders 247, 265–266, 336 reactive seizures 54–95 amphetamine and amphetamine-like
compounds 92–93 animal-related poisoning 88–89 automotive products 82–84 exogenous toxic disorders 70–73 Guarana and ma huang 94–95 HE see hepatic encephalopathy (HE) hexachlorophene 84 5-HTP 94 hypernatraemia 64–66 hypocalcaemia 66–67 hypoglycaemia 55–56 hyponatraemia 63–64 insecticides 73–77 insulinoma 56–58 ivermectin and macrocyclic
lactones 89–91 lead 84–85 levamisole 91 metabolic disorders 54–55 metaldehyde 77–78 methylxanthines 91–92 metronidazole 89 mycotoxins, penitrem A and
roquefortine 87–88 nutritional disorders 67–70 poisonous plants 85–87 prevalence 54 renal-associated encephalopathy 62–63 rodenticides 78–82 SSRIs 93–94 toxic disorders 54
rectal benzodiazepines, CS 507–509 canine studies 507–508 concentrations 507 diazepam 507 in human studies 508 lipid-soluble drugs 508
Index 589
midazolam 509 pilot studies 508–509
refractory seizures 28– 34 AEDs 28 brain structure and function 28 epilepsy 28 estimation, dog population 28 pharmacoresistant epilepsy
clinical risk factors 30–31 definition 29–30 genetic risk factors 30 mechanisms 31–34
pseudoresistance 31 treatment failure 28– 29
refractory status epilepticus (RSE) 529–532 anaesthetic drugs 529 barbiturates 531 continuous benzodiazepine infusion 529–530 inhalational anaesthesia 531 ketamine 531 lacosamide (LCM) 531–532 levetiracetam 530 propofol 530–531 resistant seizure activity 529
REM sleep disorders see rapid eye movement
(REM) sleep disorders renal-associated encephalopathy 62–63 rodenticides 78–82
bromethalin 79–80 sodium monofluoracetate 81–82 strychnine 78–79 zinc phosphide 80–81
RSE see refractory status epilepticus (RSE)
rufinamide 470–473 adverse effects 472 dosing and monitoring 472 drug–drug interactions 471, 473 efficacy 472–473 mechanism of action 464, 471 metabolism and pharmacokinetics 471 molecular structure 470–471 neuronal receptor targets 464
S-adenosyol l -methionine (SAMe) 62, 384 salivary therapeutic monitoring 368–369 SE see status epilepticus (SE) seizure patient
aetiologic diagnosis 274 compassion and empathy 275 EEG abnormalties 274 neurological examination 275–276 physical examination 275 signalment 274–275
seizures and epilepsies classification 39–51 definition 39 human medicine 40–42 electroclinical syndromes 40 generalized seizures 40– 41 idiopathic 40 ILAE proposal, 1981, 1989 and
2010 40–42 ictus 39 prodromal phase 39
selective serotonin reuptake inhibitors (SSRIs) 93–94
serum phenobarbital concentrations 387–388 AEM 387 cytopenias 381 daily dosage 387 dosage modulation 378 hepatotoxicity 387 superficial necrolytic dermatitis 381
sleep disorders 264–266 narcolepsy/cataplexy see narcolepsy REM 265–266 and tremor syndromes 244
SNEs see subcutaneous needle
electrodes (SNEs) sodium monofluoracetate 81–82 spinal nerve reflexes 275, 293–296
cutaneous trunci 294–295 extensor carpi radialis reflex 293 flexor 294 patellar reflex 294 perineal 294
spongy degenerations 49, 187, 194
status epilepticus (SE) 519–533 AEM treatment 526–529 body systems affected 519, 520 cats 522 clinical features 523 continuous EEG monitoring 532–533 definition 519 dogs 521–522 GABAA receptors 19 goals of treatment 524 identification and treatment, causal
factors 524–525 brain imaging procedures 524– 525 cytotoxic and vasogenic oedema 525 diagnostic investigation 524 hypoglycaemia and
laboratory blood tests 524 LCM 467–468 maintain vital functions 525–526
intravenous access 526 management of patient 525 oxygenation, airway and patient
temperature regulation 526 medications dosages 526 mortality rate 522
status epilepticus (SE) (continued ) pathophysiology 519–521 prognosis 522 rodent brains 12 RSE 529–532 survival rates 522–523 systemic features 523 transition to maintenance therapy 532
strychnine 78 subcutaneous needle electrodes (SNEs) 329 subdermal wire electrodes (SWEs) 329 sudden unexpected death in epilepsy
(SUDEP) 231 SUDEP see sudden unexpected death in epilepsy (SUDEP)
superficial necrolytic dermatitis 381–382 footpad lesions 381, 382 hepatic catabolism 382 rostral muzzle 382 skin disease 381
surgical therapy 550–553 categories 550 complications 550–551 hemispheric epilepsy syndromes 552 lesional neocortical epilepsy 552 MTLE 551–552 ‘non-lesional’ neocortical epilepsy 552 patient’s cognitive and neurologic status 551 risks 551 in veterinary patients 552–553
SWEs see subdermal wire electrodes (SWEs)
symptomatic/structural epilepsy antiepileptic medications and treatment 101 CD see canine distemper (CD) CNS 112–115, 123–147, 159–167
bacterial see bacterial CNS infections hydrocephalus 159, 162–167 inflammatory/infectious
see inflammatory CNS disease
malformations, forebrain 159–162 CVA see cerebrovascular accidents (CVAs) description 101 disorders, degenerative see degenerative
diseases FIP 121–123 focal lesions 101 forebrain disorder 101 GME see granulomatous
meningoencephalomyelitis (GME) neoplasia see neoplasia prevalence 101 TBI 152–159
syncope approach, patient 263–264 and bradyarrhythmias 554 cardiovascular disease 248 description 261
pathophysiology 261–262 treatment 264
thiamine/vitamin B1 deficiency 67–70 activation 67–68 clinical signs 70 diagnosis
focal haemorrhagic necrotic lesions 69, 70 high-performance liquid
chromatography 70 pathologic changes 70 presumptive 70 treatment 70 T2weighted and FLAIR images 68–70
outcomes 67 TIA see transient ischaemic attack (TIA) TMS see transcutaneous magnetic
stimulation (TMS) toad 88–89 topiramate 458–461
adverse reactions 460 antiseizure medication 458 dosing and monitoring 460 drug interactions 460 efficacy 461 mechanism of action 458, 459 metabolism 459–460 molecular structure 458, 459 pharmacokinetics 458–459
transcutaneous magnetic stimulation
(TMS) 541–543 transient ischaemic attack (TIA) 102 traumatic brain injury (TBI) 152–159
adult rodent brain 13 diagnostic investigations 156–158 dogs and cats 152 emergency treatment 153, 154 epidemiology 224 initial assessment 153 intracranial haemorrhage 156–158 and MGCS 153, 154 monitoring parameters 153, 154 pathophysiology 152–153 post-traumatic seizures and
epilepsy 153, 155–156 AEMs 156 dogs and cats 155–156 early and late 153, 155 impact 153 intractable 155 pathogenesis 155 risk factors 155
Index 591
vagus nerve stimulation (VNS) 537–541 clinical human use 538 CS 512–513 description 537–538 efficacy 539–540 magnet-activated stimulus parameters 538 mechanism of action 539 non-invasive VNS 541 side effects and safety 540–541 stimulation, cervical portion 538 stimulator settings 538–539 surgical implantation procedure 538
v-EEG see video EEG (v-EEG) vestibular ataxia 284, 285, 296 vestibulo-ocular reflex 114, 154, 289 veterinary medicine 42–51
aetiology-based classification 46–47 idiopathic/primary epilepsy 49–50 parallelism with ILAE 2010
proposal 46–49 precipitated seizures 50 probable symptomatic/cryptogenic 49 reactive seizures 46 reflex seizures 50–51 structural/secondary
epilepsy 46–47, 49 anti-epileptic medication response 213 diazepam 484 drug trials 29 efficacy, AEMs 363 electroencephalography (EEG) 318 encephalitis-associated seizures 113 felbamate 456 focal-onset seizures 44– 46
abnormal neuronal activity 44 autonomic 44 complex and simple 45 feline 46 ILAE classification 44 location 45 motor 44 partial 44 report, cats and dogs 45–46 secondarily generalized 45 sensory 44–45 stereotyped 45
generalized-onset seizures 40–44 absence 44 cerebral hemispheres 42
clonic 43 myoclonic 43–44 primary 42 tonic 43 tonic-clonic 42–43
grading 179 guidelines 366, 368 ICP monitoring 156 initial assessment and
emergency treatment 153 involuntary movement abnormalities 253 pharmacokinetic interactions 443 PTE 156 rapid eye movement sleep disorders 266 seizure freedom and quality of life 34
video EEG (v-EEG) 325, 341–342 VNS see vagus nerve stimulation (VNS)
zinc phosphide 80–81, 303 ZNS see zonisamide (ZNS) zonisamide (ZNS) 418–422
adverse effects 418–420 dose-related 418–419 idiosyncratic reactions 419–420 laboratory changes 420
anti-epileptic medication 362, 414, 422 dosing and monitoring 420 efficacy 414 and levetiracetam 378 mechanism of action 414–415 metabolism 415–417
absorption 416 cat and dogs 416 concentration and disposition 416 humans 416 parameters, dogs 416, 417 and pharmacokinetics 416
molecular structure 414, 415, 422 new generation AEMs 169, 179, 532 pharmacokinetic interactions
CYP450 isoenzymes 418 hepatic CYP3A4 416, 418 phenobarbital administration
379, 418 safety and efficacy 391 salivary therapeutics 368–369 sulfonamide-based medication 422 tolerance 414 treatment, brain disorders 151